
顶空气相色谱法测定不同品种食用植物油中的溶剂残留量
2017-03-27 21:51周玮婧江小明
湖北农业科学 2017年2期
关键词:植物油
周玮婧+江小明

摘要:建立了一种顶空-毛细管气相色谱快速测定不同品种食用植物油中溶剂残留量的方法。在80 ℃下振摇30 min后顶空进样,采用DB-624毛细管柱(0.53 μm,0.25 mm,30 m)和氢火焰检测器测定,外标法定量。结果表明,在0~500 μg范围内,六号溶剂的峰面积与其含量呈良好线性关系,相关系数为0.999,检出限为0.096 mg/kg,8种食用植物油平均加标回收率为87.5%~106.0%,相对标准偏差(RSD)为0.30%~3.05%。该方法简便快速、结果准确、重现性好,适合各种植物油中溶剂残留量的测定。
关键词:顶空气相色谱法;溶剂残留量;植物油;六号溶剂
中图分类号:O657.7 文献标识码:A 文章编号:0439-8114(2017)02-0334-05
DOI:10.14088/j.cnki.issn0439-8114.2017.02.032
食用油的加工生产通常有精炼、压榨、浸出3种方法,其中较其他方法相比,浸出法具有出油率高等优点,故倍受生产企业的青睐[1]。目前,中国常用的萃取植物油的溶剂为六号溶剂,其主要成分为六碳烷烃,如果萃取不彻底,最后获得可食用的植物油中就可能有溶剂残留的存在,不仅影响油的品质,更危害人的身体健康[2,3]。为了保证食品安全,中国强制性标准GB 2716-2005对溶剂残留进行了规定:采用浸出工艺生产的植物油的溶剂残留量为50 mg/kg,采用压榨工艺生产的植物油的溶剂残留量为未检出(压榨法中溶剂残留量≤10 mg/kg时即视为未检出)[4]。
溶剂残留分析一般采用静态顶空、动态顶空、固相微萃取等样品采集方法,经气相色谱分离后定量分析。其中,静态顶空法因其成本低、重现性好而广泛用于食品、药品、材料制品等中的溶剂残留分析。国家标准GB/T 5009.37-2003《食用植物油卫生标准的分析方法》中以该法配合填充柱,手动进样测定植物油中的溶剂残留量,但该方法柱效低,对六号溶剂的分离度差,所以检测结果准确度较低[5]。为此,建立一种快速、准确的测定植物油中溶剂残留量的方法尤为重要,本研究参照现行的国标并进行优化,建立了顶空进样,毛细管气相色谱分离,氢火焰电离检测器(FID)检测,以六号溶剂为标准溶液定量的溶剂残留方法,该法样品前处理简单、分析时间短、回收率和精密度较高,且结果可靠、检出限低,适合植物中溶剂残留量的快速测定。
1 材料与方法
1.1 材料与试剂
基体植物油:与被检测样品同品种的无残留溶剂的植物油;六号溶剂标准溶液(国家粮食局科学研究院)浓度为10 mg/mL,溶剂为N,N-二甲基乙酰胺;顶空瓶:Agilent公司20 mL顶空瓶及聚四氟乙烯瓶垫,压盖器等;注射器:Agilent公司气相色谱微量进样针。
1.2 仪器与设备
Agilent 7890B型气相色谱仪:配备毛细管柱分流/不分流进样系统,FID检测器;Agilent 7697A型顶空进样器和数据处理系统,电子分析天平。
1.3 试验条件
1.3.1 色谱条件 谱柱DB-624毛细管色谱柱(0.53 μm,0.25 mm,30 m),载气氮气(99.999%),流速4.0 mL/min,进样口温度220 ℃,分流比1∶10;检测器(FID)200 ℃,氢气30 mL/min,空气400 mL/min;进样体积1 mL。柱温箱程序升温初始温度70 ℃,保持8 min,升温速率40 ℃/min,最终温度200 ℃。
1.3.2 顶空条件 载气氮气,顶空瓶口充气压125 kPa,顶空瓶口加热温度80 ℃,定量管温度85 ℃,传输气路温度90 ℃,平衡时间30 min;顶空瓶口充气增压时间0.13 min,顶空瓶口向定量管充气时间0.15 min,定量管平衡时间0.15 min,六通进样阀进样时间1 min。
1.4 标准溶液的配制及含量测定
分别称量5 g(精确到0.000 1 g)与待分析样品相同基质的5个空白基体植物油样品,放入顶空进样瓶中。用微量注射器分别迅速加入六号溶剂标准溶液(1、5、10、25、50 μL),含量分别为10、50、100、250、500 μg,用铝帽和密封垫密封后,小心涡旋振荡密封瓶,使物质充分混合。将配置好的标准溶液上机分析。称取5 g(精确到0.000 1 g)不同品种油样,在与绘制标准曲线相同的色谱条件下进行测定,根据标准曲线,计算出样品中残留溶剂的含量,试样中溶剂残留的含量按下式计算。
X=■
式中,X为试样中溶剂残留的含量,单位为mg/kg;m1为由标准曲线求得试样溶液中溶剂残留的浓度,单位为μg;m2为试样质量,单位为g。
2 结果与分析
2.1 六号溶剂组分定性分析
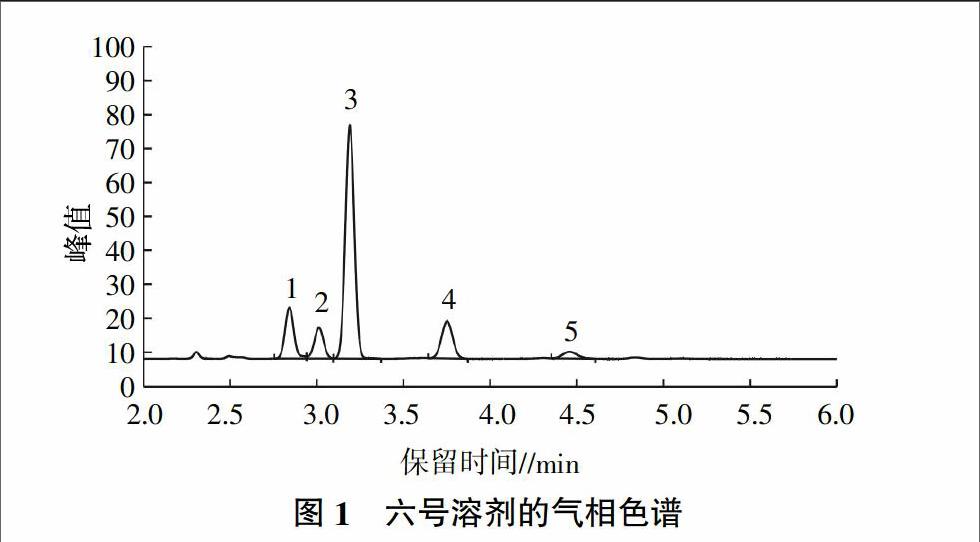
国家标准中使用的标准物质为六号溶剂,按设定色谱条件对六号溶剂标准溶液进行分析,得到气相色谱见图1。六号溶剂的5种主要组分保留时间分别为2.844、3.015、3.192、3.754和4.466 min,且各峰峰型良好。六号溶剂是以C6~C8为主的烃类混合物,包括烷烃、烯烃、环烷烃和芳香烃,经质谱定性分析发现,含有2-甲基戊烷(1号峰)、3-甲基戊烷(2号峰)、正己烷外(3号峰)、甲基环戊烷(4号峰)、环己烷(5号峰)等5种主要组分,也能检测出3-甲基己烷、正戊烷、正庚烷等微量组分。
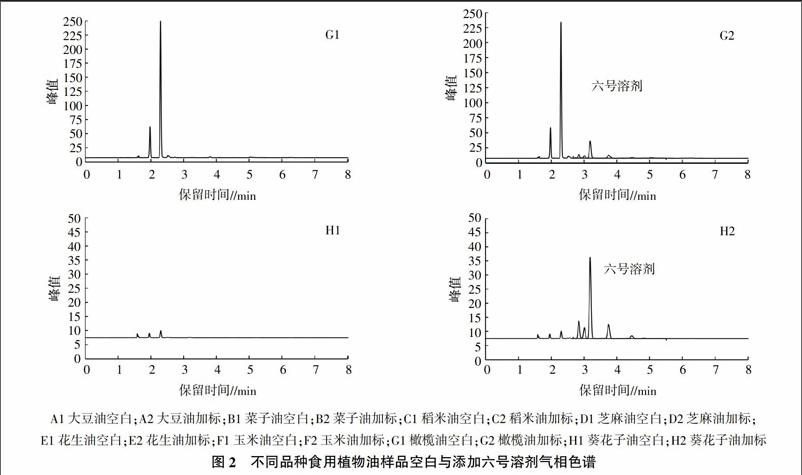
选取大豆油、菜子油、稻米油、芝麻油、花生油、玉米油、橄榄油、葵花子油等8种不同品种无溶剂残留的植物油作为基体油,并添加10 mg/kg的六号溶剂标准溶液,按设定色谱条件进行分析,结果如图2所示。在8種食用植物油中,六号溶剂5种主要成分出峰时间为2.67~5.50 min,空白样品出峰时间主要在2.67 min之前,即六号溶剂出峰之前,且各色谱峰存在明显差异,这些色谱峰为不同品种食用植物油的低沸点风味物质,其中橄榄油的风味物质浓度较高、菜子油、芝麻油的风味物质较多较杂,大豆油的风味物质较少较单一,而在该色谱条件下这些风味物质均已和六号溶剂色谱峰完全分离,说明该方法适用于大部分食用植物油的分析。
2.2 标准曲线、线性范围及检测限的测定
文献[6]报道,分析测定植物油中残留溶剂时,同种植物源植物油标准曲线可以通用,如果测定不同植物源植物油中残留溶剂,选择标准曲线的基质时应结合实际测量的要求,选择适当基质,以尽量减少误差。所以通常选用低沸点风味物质较少,空白色谱图基线较平稳的大豆油为基底油(图2)。以相应六号溶剂含量(μg)为横坐标,六号溶剂的5个主要峰面积(Pa)加和为纵坐标,绘制标准曲线。按照2.67~5.50 min出峰面积加合的方式计算,回归方程为Y=3.982X+15.22,相关系数为0.999。该法线性范围较宽,浓度在0~500 μg范围内呈线性关系良好,该方法简便快速,灵敏度高,适于对大批量样品的分析。以基体植物油为空白,加入低浓度六号溶剂标准,按“1.3”试验条件上机测定,以3倍的噪声峰高为检出限,以10倍噪声峰高为定量限。取5.00 g样品,在本试验条件下六号溶剂的检出限为0.096 mg/kg,定量限为0.32 mg/kg,符合溶剂残留检测要求。
2.3 不同品种食用油回收率与精密度的测定
在设定的色谱条件下,选取空白植物油(大豆油、菜子油、稻米油、芝麻油、花生油、玉米油、橄榄油、葵花子油)等8种不同品种植物油为基质,进行加标回收率试验。准确称取空白底油样品5 g,分别添加低(2 mg/kg)、中(10 mg/kg)、高(20 mg/kg) 3个浓度标准溶液,每个浓度平行做5次,按上述方法进行测定。由表1可见,低浓度组六号溶剂的加标回收率为87.5%~106.0%,中浓度组的加标回收率为92.4%~98.8%高浓度组六号溶剂的加标回收率为93.5%~99.5%,均能满足微量分析的要求。同时这8种食用植物油的测量精密度(相对标准偏差RSD)在0.30%~3.05%之间,均小于5%。试验结果表明,该方法准确可靠,重现性良好,满足微量分析的要求,由于测试样品覆盖了植物性食用油的各类样品,从回收率看,该方法基本能够覆盖市面上的各类食用植物油。
2.4 与国标方法测定六号溶剂的结果比较
从同一油样中分别各取7份样品,分别使用顶空-毛细管柱气相色谱仪法和国标使用的填充柱气相色谱仪法分别进行测定其六号溶剂残留量,结果见表2。用F检验和t检验分析统计两种不同方法测定结果是否有差异,结果见表2。统计量F=1.83小于显著水平α=0.05的F临界值5.05,两种测定方法结果方差无显著差异;统计量t=1.08小于显著水平α=0.05的t双尾临界值为2.26,即两种不同分析方法结果无差异。但由于填充柱的柱效低于毛細管柱,对六号溶剂的分离度差,会将样品中一些低沸点风味物质的色谱峰包裹在六号溶剂中,因而影响了测定结果。
3 结论
在现有国标方法的基础上做出了改进,采用静态顶空设备进样,DB-624毛细管色谱柱,气相色谱-火焰离子化检测器进行分析,对样品处理过程中的平衡温度和平衡时间进行优化后,选用了8种代表性的食用植物油作为基底油进行方法学分析,建立了顶空-气相色谱法测定植物油中溶剂残留的方法,大大提高了检测结果的灵敏度和准确度,该法样品前处理、分析时间短、回收率和精密度高,且结果可靠、检出限低等优点,适合植物中溶剂残留量的快速测定。目前市场上仍然存在着以浸出油和压榨油调和后的食用油冒充压榨食用油的现象,该方法的建立为植物油中残留溶剂控制提供检测依据,将为食品安全监管部门及相关食品安全工作者提供了有益的技术支持[7]。
参考文献:
[1] 王竹天,杨大进,吴永宁.食品卫生检验方法(理化部分)注解[M].北京:中国质检出版社,2008.
[2] 汪海峰,鞠兴荣,杨晓蓉,等.食用植物油中残留溶剂的高温顶空气相色谱分离与测定[J].食品科学,2006,27(2):235-238.
[3] 刘 岚.溶剂残留量对食用油的影响[J].农产品加工(综合刊),2011(2):30-31.
[4] 孙海燕,李云雁.气相色谱法测定食用油中的残留溶剂[J].粮油加工,2010(5):15-17.
[5] 周 洲,张 中,张榴萍,等.植物油中溶剂残留量测定影响因素探讨[J].食品工业,2014(1):183-186.
[6] 郑月明.色谱技术在食用植物油品质评价中的应用[D].北京:中国农业科学院,2013.
[7] 曾文锦.顶空气相色谱毛细管柱法测定浸出食用油溶剂残留量色谱条件的探讨[J].化工管理,2015(21):49-50.
猜你喜欢
公民与法治(2020年14期)2020-08-24
药学研究(2015年11期)2015-12-19
中国塑料(2015年3期)2015-11-27
邵阳学院学报(自然科学版)(2015年2期)2015-06-05
华东理工大学学报(自然科学版)(2014年2期)2014-02-27
中国粮油学报(2014年7期)2014-02-06
食品科学(2013年6期)2013-03-11
