
结核分枝杆菌北京基因型菌株大片段的多态性研究
2018-03-07 02:46谢彤孙蕊巨韩芳王春花穆成王志锐赵慧
中国防痨杂志 2018年3期
谢彤 孙蕊 巨韩芳 王春花 穆成 王志锐 赵慧
结核病的分子流行病学研究提示,受环境和宿主因素的影响,全球不同地区流行的主要结核分枝杆菌(Mycobacteriumtuberculosis,MTB)的主要基因型并不相同。在所发现的MTB基因型中,北京基因型的流行在全球传播的范围最广[1]。北京基因型MTB菌株与其他基因型菌株相比,该基因型在低流行区更容易在人群中发生近期传播,从而导致感染该基因型的结核病患者比例呈逐年升高的趋势[2]。北京基因型MTB较其他基因型菌株更容易产生耐药,而产生的耐药菌株又促进了北京基因型MTB在人群中的传播,因此,在有些地区的流行病学研究显示,北京基因型与耐药结核病、特别是耐多药结核病(multidrug-resistant tuberculosis,MDR-TB)的传播密切相关[3]。我国是北京基因型MTB的高流行区,特别是在北方地区,大约90%临床分离的MTB菌株属于北京基因型,是全球已报道北京基因型MTB流行最高的地区[4]。
对基因组分析证实,北京基因型MTB在进化过程会发生插入序列6110(IS6110)的转位和特异性的差异区域(region of difference,RD)缺失。根据该基因型菌株基因组NTF(noise transfer function)区域中IS6110是否存在,北京基因型被分为古代与现代两个亚型[5]。北京基因型古代株的NTF区中没有IS6110,而北京基因型现代株NTF区中至少含有1个IS6110。此外,不同的北京基因型菌株在进化过程中还会出现不同的RD片段缺失,由此产生菌株大片段多态性。依据不同MTB菌株基因组中RD181、RD150以及RD142的缺失情况,北京基因型被分为5个进化分支[6]。笔者对天津地区临床实验室分离培养获得的北京基因型MTB菌株基因组中RD片段的多态性与NTF区中IS6110进行分析,以期揭示在天津地区流行的北京基因型菌株所形成的各个分支及其流行的状况。
材料和方法
1.菌株来源:选取天津市结核病参比实验室收集的2014年1月至2016年6月从天津地区结核病患者痰标本中分离培养的北京基因型MTB,共567株。其中,308株分离培养自天津市区(和平区、河东区、河西区、南开区、河北区和红桥区)结核病患者,221株分离培养自滨海新区(包括塘沽、汉沽、大港)、环城区(包括东丽、西青、津南、北辰)及其他区县(包括武清、宝坻、宁河、蓟州),38株分离培养自监狱局系统结核病患者。所有菌株经对硝基苯甲酸试验和噻吩-2-羧酸肼培养基生长试验,排除非结核分枝杆菌和牛分枝杆菌。H37Rv标准株购自中国药品生物制品检定所。
2. MTB基因组提取:临床分离培养的MTB菌落转移至装有450 μl磷酸盐缓冲液(PBS)的1.5 ml Eppendorf管中,细菌经80 ℃、60 min灭活与溶菌酶消化后,采用十六烷基三甲基溴化铵法(CTAB法)提取菌株的基因组DNA[7]。提取后的基因组DNA溶于1×TE缓冲液中,基因组提取所需试剂均购自美国Sigma公司。
3. 北京基因型MTB鉴定:根据北京基因型MTB的基因组中RD207缺失这一特征,通过多重PCR试验分析菌株基因组在Rv2616至Rv2819区域的缺失情况。在PCR反应体系中同时加入2对引物,分别针对北京基因型基因组中的Rv2820区域和北京基因型菌株基因组中缺失的Rv2819区域。25 μl PCR反应体系中包括2×Taq Master-Mix 12.5 μl(购自北京天根生化科技有限公司)、终浓度为0.4 μmol/L的引物及10~50 ng的菌株基因组DNA。若MTB为北京基因型,则PCR产物扩增片段长度为393 bp;而非北京基因型PCR扩增产物片段长度为570 bp[7]。
4.基因组NTF区域IS6110分析:北京基因型菌株基因组NTF 区IS6110 插入序列的分析采用Wada等[8]报道的方法。使用上游引物MDR-6与下游引物MDR-6r分析NTF 区是否存在IS6110,如果含有IS6110,则为现代株,其扩增产物为1.5 kb;反之则为古代株,其扩增产物为302 bp。
5.RD多态性分析:采用多重PCR分别分析北京基因型菌株基因组中RD105、RD181、RD150与RD142差异区域的缺失情况,根据PCR反应扩增产物的长度确定RD序列的缺失或完整,引物序列参考Reed等[9]的报道。在50 μl PCR反应体系中含有25 μl的Q5 Hight-Fidelity 2×Master-Mix Taq酶(购自北京NEB有限公司),2.4 μl 10 μmol/L的引物,以及10~50 ng的基因组DNA。PCR反应条件为95 ℃ 变性5 min;94 ℃ 30 s,60 ℃ 30 s,72 ℃ 40 s,30个循环;72 ℃反应10 min。
结 果
1.基本情况:567株MTB临床分离株中,517株为北京基因型,占91.2%,是绝对的优势菌株。对517株北京基因型菌株NTF区IS6110分析,70株(13.5%)为古代株,即菌株的NTF区不含有IS6110;447株(86.5%)为现代株,其基因组NTF区中存在IS6110。
2.基于大片段多态性的进化分支:517株北京基因型菌株根据基因组中NTF区IS6110的存在情况和RD的多态性被分为5个进化分支(表1)。本研究中所有的北京基因型菌株基因组中均发生RD105缺失。基因组中存在RD181片段的RD181(+)菌株被认为是北京基因型早期进化分支,在所有的北京基因型菌株中RD181(+)菌株为22株(4.3%),RD181(-)的北京型菌株为495株(95.7%)。RD181(-)菌株中,基因组NTF区无IS6110的北京基因型古代株为41株,占全部北京基因型菌株的7.9%。22株RD181(+)的菌株中,其基因组NTF区均无IS6110序列,即均为北京基因型古代株。在447株北京基因型现代菌株中,404株基因组中存在完整的RD150序列,占全部北京基因型菌株的78.1%;43株为RD150缺失的北京基因型现代株,占全部北京基因型菌株的8.3%,占全部北京基因型现代株的9.6%(43/447)。70株北京基因型MTB古代株中,同样发现有7株基因组发生RD150缺失,占全部北京基因型古代株的10.0%(7/70)。由此可见,根据大片段多态性分析天津地区流行的北京基因型MTB被分为5个分支,其中RD181(-)/RD150(+)的北京基因型现代株所占的比率最高,是北京基因型的主要流行分支(图1)。RD142差异序列存在于所有分析的北京基因型菌株基因组中。
讨 论
MTB在长期进化过程中其基因组会发生大片段的转位插入或缺失,而这一变化可能会影响菌株的毒力和宿主对MTB的免疫,使某些菌株更容易在人群中传播流行,在这一地区形成优势菌株,造成全球不同区域流行的MTB主要基因型也各不相同。全球范围内,不同地区进化出至少6个基因型,每个基因型的MTB基因组中都存在特异性的RD缺失序列。因此,通过分析菌株基因组中这些特异性的RD缺失序列,可以鉴定MTB所属的基因型[10-11]。对从99个国家和地区流行的北京基因型MTB的分子流行病学研究发现,该基因型大约在6600年前起源于包括我国东北在内的东亚地区,随着近代大规模人口迁徙的路径逐渐扩散至全球各地[12]。北京基因型MTB的重要特征为RD207和RD105片段的缺失。虽然Rindi等[13]的研究发现,极少数北京基因型MTB基因组中含有RD105差异序列,但在我国流行的北京基因型菌株基因组中还未发现含有RD105的报道,因此检测RD207和RD105缺失情况可用于鉴定北京基因型MTB。
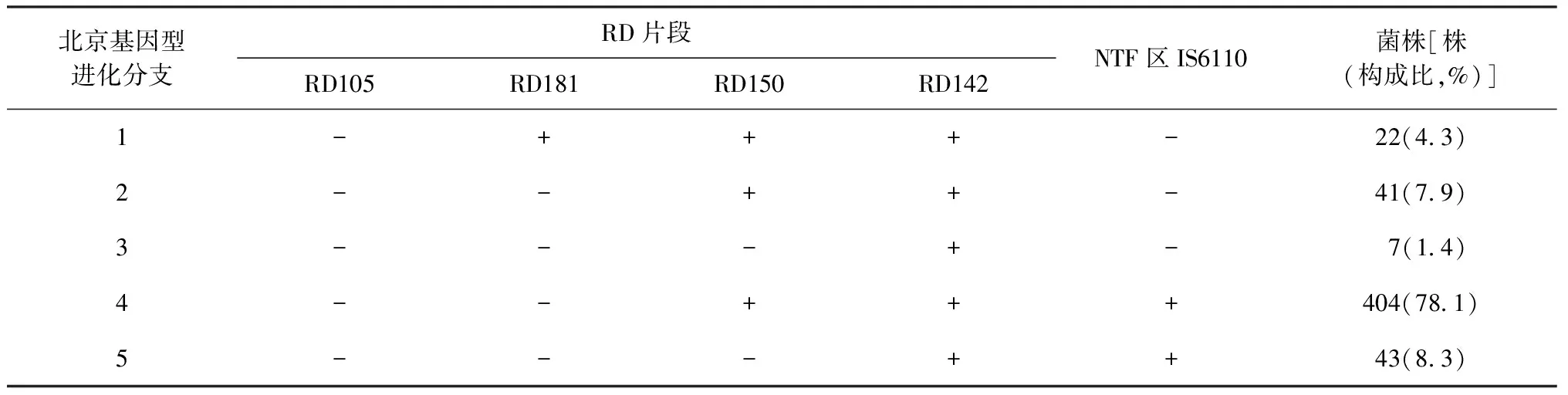
表1 517株5个分支北京基因型结核分枝杆菌菌株大片段多态性分析结果
注“+”:表示完整;“-”:表示缺失
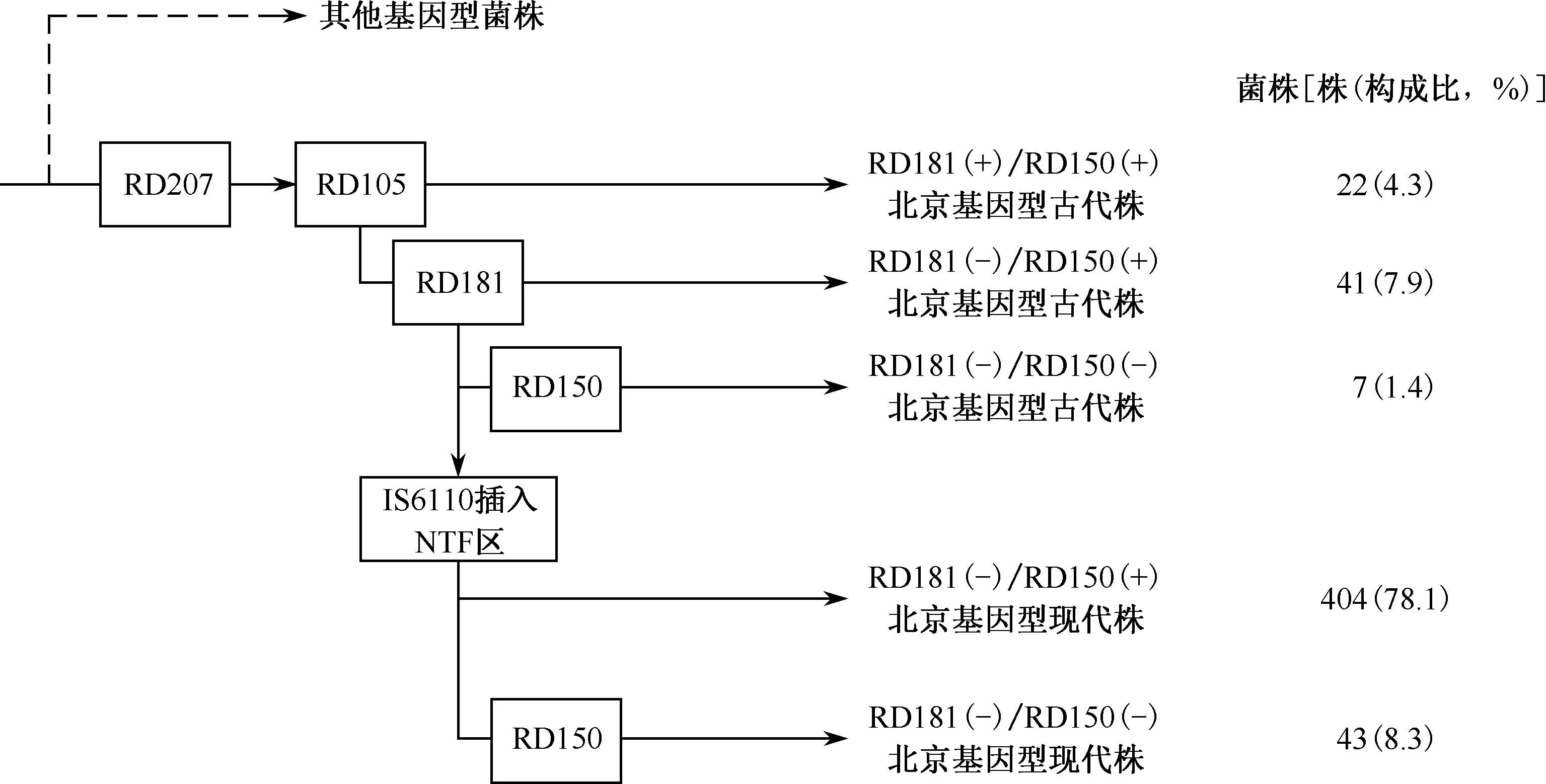
矩形框中表示在菌株进化过程中RD序列的缺失和IS6110在NTF区的插入图1 北京基因型结核分枝杆菌进化途径和所形成的进化分支
根据 RD片段多态性研究提出的北京基因型进化途径中,MTB菌株首先发生RD207缺失进化成为北京基因型,随后几乎所有的北京基因型菌株发生RD105缺失。在以后的持续进化过程中不同的菌株按顺序可能出现RD181、RD150和RD142的缺失[13]。本次研究显示,RD181缺失的北京基因型菌株为495株,占95.7%,高于我国东部地区。对东部地区不同省市结核病患者临床分离北京基因型菌株的两项流行病学研究中,RD181(-)菌株仅占全部北京基因型菌株的67.8%和87.0%,提示北京基因型各个进化分支的菌株在我国不同地区的流行情况存在差异[14-15]。根据基因组NTF区中IS6110的存在与否,将北京基因型菌株分为古代株(IS6110缺失)和现代株(IS6110存在)。本研究的菌株中北京基因型的现代株为447株(86.5%),与文献报道我国东部地区现代株所占全部北京基因型菌株的87.1%相近[15]。
基于RD片段的多态性和IS6110在NTF区的插入情况,本次研究中的517株北京基因型MTB被分为5个分支,其中RD181(-)/RD150(+)的北京基因型现代株这一分支所占的比率最高(78.1%)。22株RD181(+)的菌株全部为北京基因型古代株,提示北京基因型菌株在进化过程中首先发生RD181的缺失,然后才出现IS6110在NTF的插入,进化顺序与其他研究报道的结果一致[13,16]。本次研究发现7株RD150(-)的北京基因型古代株,这是基于大片段多态性分析北京基因型MTB进化研究中首次发现的进化分支,同时研究数据显示RD150(-)的菌株在北京基因型MTB古代株和现代株的比例非常接近,分别占10.0%(7/70)和9.6% (43/447),提示RD150大片段的缺失并不仅出现在北京基因型现代株中,而是在菌株进化成为现代株与古代株2个分支后,这2个分支的MTB在传播过程中均有少量的菌株(大约10%)进一步发生RD150的缺失。笔者对基因组中大片段多态性分析表明,北京基因型菌株的进化路径为MTB首先发生RD207和RD105缺失进化成为北京基因型,随后出现RD181片段的缺失,然后发生IS6110序列在NTF区的插入形成现代北京基因型菌株;少量(大约10%)的古代北京基因型和现代北京基因型菌株在随后的进化过程中出现RD150的缺失。Tsolaki等[6]的研究发现部分北京基因型菌株可能会出现RD142缺失。笔者的研究结果显示北京基因型菌株基因组均含有完整的RD142序列,其他一些国内外关于北京基因型进化的研究中,同样也未发现RD142缺失的MTB[13-14],提示RD142差异片段在北京基因型菌株中的多态性较低。
虽然在少数国家流行的北京基因型MTB以古代株为主,但在我国和全球的大部分地区北京基因型现代株的流行更为广泛[12]。分子流行病学调查证实,北京基因型现代株容易在人群发生近期传播和暴发流行。这一北京基因型分支更容易流行的机制还未完全阐明,与北京基因型古代株相比,RD150(+)的北京基因型现代菌株在诱导巨噬细胞产生免疫应答时,巨噬细胞释放各类细胞因子水平明显降低[17]。小鼠感染实验证实RD150(+)北京基因型现代株较北京基因型古代株会对肺组织造成更大的病理损伤,提示这一进化分支具有更强的毒性[18]。此外,大规模的人群流行病学研究提示,北京基因型现代株较该基因型的古代株更容易逃避卡介苗接种所产生的免疫反应,因此卡介苗的接种可能会促进北京基因型现代株在人群中的传播[19]。上述的研究提示,鉴于天津地区流行的北京基因型分支为RD150(+)的现代株,而这一流行特点会为天津地区结核病的预防和治疗工作提出更大的挑战。同时本研究结果也说明,非常有必要在我国的其他地区开展对北京基因型各个亚型菌株流行的监测工作。
[1] Ramazanzadeh R, Sayhemiri K. Prevalence of Beijing family inMycobacteriumtuberculosisin world population: systematic review and meta-analysis. Int J Mycobacteriol, 2014, 3(1): 41-45.
[2] Garzelli C, Lari N, Rindi L. Genomic diversity ofMycobacteriumtuberculosisBeijing strains isolated in Tuscany, Italy, based on large sequence deletions, SNPs in putative DNA repair genes and MIRU-VNTR polymorphisms. Tuberculosis, 2016, 97: 147-153.
[3] Hanekom M, Gey van Pittius NC, McEvoy C, et al.MycobacteriumtuberculosisBeijing genotype: a template for success. Tuberculosis (Edinb), 2011, 91(6): 510-523.
[4] Wan K, Liu J, Hauck Y, et al. Investigation onMycobacteriumtuberculosisdiversity in China and the origin of the Beijing clade. PLoS One, 2011, 6(12): e29190.
[5] Mokrousov I, Ly HM, Otten T, et al. Origin and primary dispersal of theMycobacteriumtuberculosisBeijing genotype: clues from human phylogeography. Genome Res, 2005, 15(10): 1357-1364.
[6] Tsolaki AG, Gagneux S, Pym AS, et al. Genomic deletions classify the Beijing/W strains as a distinct genetic lineage ofMycobacteriumtuberculosis. J Clin Microbiol, 2005, 43(7): 3185-3191.
[7] 赵德福, 谢彤, 巨韩芳, 等. 利用多重PCR方法快速鉴定结核分枝杆菌北京基因型菌株. 中国防痨杂志, 2010, 32(6): 315-317.
[8] Wada T, Iwamoto T, Maeda S. Genetic diversity of theMycobacteriumtuberculosisBeijing family in East Asia revealed through refined population structure analysis. FEMS Microbiol Lett, 2009, 291(1): 35-43.
[9] Reed MB, Pichler VK, McIntosh F, et al. MajorMycobacteriumtuberculosislineages associate with patient country of origin. J Clin Microbiol, 2009, 47(4): 1119-1128.
[10] Gagneux S, Small PM. Global phylogeography ofMycobacteriumtuberculosisand implications for tuberculosis product development. Lancet Infect Dis, 2007, 7(5): 328-337.
[11] Faksri K, Hanchaina R, Sangka A, et al. Development and application of single-tube multiplex real-time PCR for lineage classification ofMycobacteriumtuberculosisbased on large sequence polymorphism in Northeast Thailand. Tuberculosis (Edinb), 2015, 95(4): 404-410.
[12] Merker M, Blin C, Mona S, et al. Evolutionary history and global spread of theMycobacteriumtuberculosisBeijing lineage. Nat Genet, 2015, 47(3): 242-249.
[13] Rindi L, Lari N, Cuccu B, et al. Evolutionary pathway of the Beijing lineage ofMycobacteriumtuberculosisbased on genomic deletions and mutT genes polymorphisms. Infect Genet Evol, 2009, 9(1): 48-53.
[14] Liu M, Jiang W, Liu Y, et al. Increased genetic diversity of theMycobacteriumtuberculosisW-beijing genotype that predomi-nates in eastern China. Infect Genet Evol, 2014, 22: 23-29.
[15] Hu Y, Mathema B, Zhao Q, et al. Comparison of the socio-demographic and clinical features of pulmonary TB patients infected with sub-lineages within the W-Beijing and non-BeijingMycobacteriumtuberculosis. Tuberculosis (Edinb), 2016, 97: 18-25.
[16] Maeda S, Wada T, Iwamoto T, et al. Beijing familyMycobacteriumtuberculosisisolated from throughout Japan: phylogeny and genetic features. Int J Tuberc Lung Dis, 2010, 14(9): 1201-1204.
[17] Chen YY, Chang JR, Huang WF, et al. The pattern of cytokine production in vitro induced by ancient and modern BeijingMycobacteriumtuberculosisstrains. PLoS One, 2014, 9(4): e94296.
[18] Ribeiro SC, Gomes LL, Amaral EP, et al.Mycobacteriumtuberculosisstrains of the modern sublineage of the Beijing family are more likely to display increased virulence than strains of the ancient sublineage. J Clin Microbiol, 2014, 52(7): 2615-2624.
猜你喜欢
传染病信息(2022年4期)2022-11-23
河南医学研究(2022年18期)2022-09-30
西部医学(2022年9期)2022-09-26
世界科学技术-中医药现代化(2022年3期)2022-08-22
黑龙江大学自然科学学报(2022年1期)2022-03-29
昆明医科大学学报(2022年2期)2022-03-29
计算机系统应用(2021年10期)2022-01-06
昆明医科大学学报(2021年6期)2021-07-31
昆明医科大学学报(2021年3期)2021-07-22
学生天地(2019年28期)2019-08-25
