
氢气及壬基蒽在铂团簇上吸附活化的电子作用机制初探
2018-08-10 09:48王春璐赵晓光王丽新叶蔚甄代振宇
石油学报(石油加工) 2018年4期
王春璐, 任 强, 赵 毅, 赵晓光, 王丽新, 叶蔚甄, 代振宇, 周 涵, 龙 军
(中国石化 石油化工科学研究院, 北京 100083)
多环芳烃主要存在于石油高沸点(>350℃)馏分中,如石油的减压馏分油(VGO)及更重的馏分中,此外在二次加工重油如焦化蜡油(CGO)、催化裂化轻循环油(FCC-LCO)中多环芳烃也有很高占比。刘颖荣等[1]对不同原油多个馏分段中的多环芳烃进行GC-MS表征发现,随馏分逐渐变重,三环芳烃在馏分油中所占的比重逐渐增加,是一类比较有代表性的多环芳烃。2016年环境保护部、国家质检总局联合发布《轻型汽车污染物排放限值及测量方法(中国第六阶段)》分步实施方案,方案所设置的国六a和国六b 两个排放限值方案将分别于2020年和2023年实施。意见稿中将车用柴油中的多环芳烃质量分数由原来的≤11% 降低为≤7%。由此可见,进一步提高多环芳烃的转化率对于改善油品品质,提高汽油、柴油、煤油等产品的收率和延长催化剂使用周期等方面都有至关重要的作用。如何能够以较低的成本、更加高效地将多环芳烃转化为高附加值的轻质油品已成为石油炼制工业亟待解决的问题之一。
反应物分子在催化剂活性位点上的化学吸附是反应物分子活化的前提,而物理吸附则很可能是化学吸附进行的重要原因[2]。芳烃在过渡金属上的吸附主要通过芳烃的π轨道与过渡金属的d轨道发生相互作用,目前已基本形成共识,然而这些研究多是基于环数较少的无取代侧链芳烃在理想体相金属表面的吸附研究[3-5],与实际反应体系尚存在一定差距,且对有H2共存的共同吸附体系研究也尚未有报道,可见提高对多环芳烃在过渡金属表面的吸附理解对于催化剂改性、提高催化加工效率都具有重要意义。
1 研究方法与模型选取
1.1 研究方法
采用Material Studio软件中基于分子动力学和蒙特卡洛原理的Adsorption Locator模块,能够获取所考察体系的初始吸附构象,并对吸附物在吸附质上能量较低的吸附位点进行搜索。具体参数设置如下:力场选用COMPASS,系综选用NPT,最大载荷步数设定为105,生产步数设定为105,电荷平衡法选用QEq。
量子力学方法以分子中电子的非定域化为基础,通过计算求解Schrödinger方程,能够研究体系的结构和电子性质,进而描述以电子主导的成键作用,适于讨论化学键的断裂或形成过程。利用Material Studio 8.0的DMol3模块进一步优化所获取的初步吸附构象,能够对吸附体系的能量、轨道、和电子结构进行更高精度的分析。具体参数设置如下:采用非局域态密度近似GGA,交换相关能函数选用BLYP,基组选用DNP,自洽迭代SCF计算的收敛阈值为2.70×10-4eV。为加速收敛过程,Thermal smearing值设为1.36×10-2eV。
1.2 模型选取
1.2.1 模型化合物选取
结合Ali等[6]对VGO馏分中多环芳烃平均结构的表征结果,及刘颖荣等[1]对多环芳烃的组成分析可知,三环芳烃是一类比较有代表性的多环芳烃。为充分考虑实际油品中多环芳烃的取代侧链可能的空间位阻作用,笔者选取带有正构壬烷基取代侧链的三环芳烃壬基蒽作为多环芳烃模型化合物,为构建侧链适宜的取代位置,分别考察了壬烷基在蒽环不同取代位置的能量。
国际纯粹与应用化学联合会(IUPAC)对稠环芳烃类物质有统一的命名与芳香碳编号规则,依照该规则蒽分子中芳香碳的IUPAC编号如图1所示。
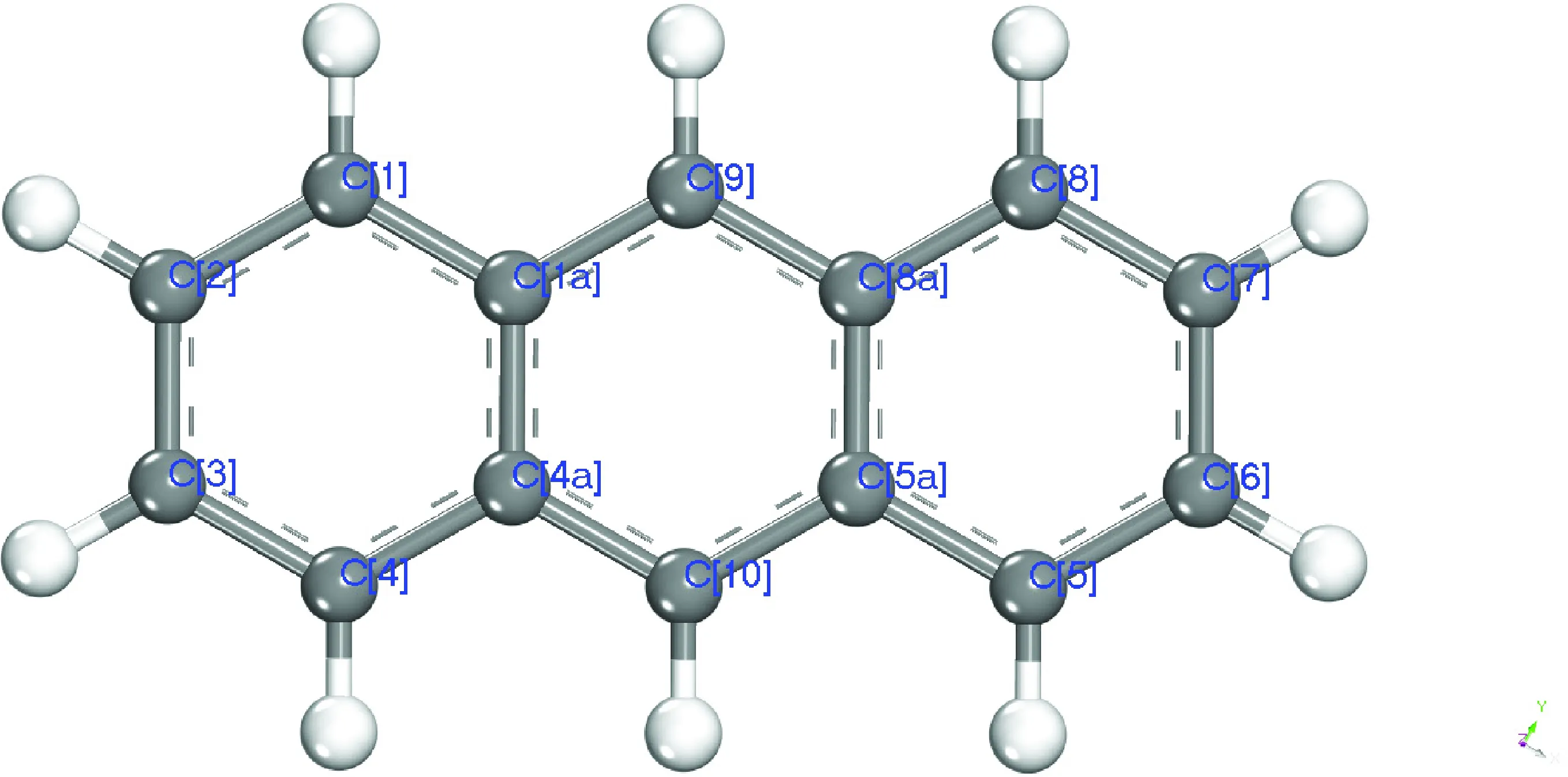
图1 蒽的分子结构及其碳原子的IUPAC编号Fig.1 Structure of anthracene and IUPAC serial numbers of carbon atoms Grey: Carbon atoms; White: Hydrogen atoms
蒽分子为对称结构,壬基取代基在蒽分子上存在3种较为可能的取代位,分别为1-壬基蒽、2-壬基蒽和9-壬基蒽。对3种取代位的结构进行优化,得到3种结构的能量如表1所示。

表1 壬基蒽结构优化能量比较Table 1 The optimized energy of nonylanthracene
对化合物来说,体系的能量越低结构稳定性越强,则在混合体系中该化合物稳定存在的可能性越高。由表1可知,9-壬基蒽的能量略低,且与另外2种结构相比空间位阻作用更大,更具有研究代表性,因此选取9-壬基蒽作为模型化合物。经模拟得到9-壬基蒽的沸点为453.6℃±12℃,其能较好地代表加氢裂化工艺所加工VGO馏分段中的多环芳烃化合物,对其他加工工艺也有一定借鉴意义。
1.2.2 加氢催化剂模型构建
加氢裂化催化剂是由加氢组分和酸性组分两个必要组分组成的双功能催化剂[7]。加氢活性主要由加氢金属提供,按照活性与价值可分为非贵金属与贵金属两大类[7]。其中非贵金属加氢裂化催化剂常用硫化态的双金属组合为加氢活性组分。根据王薇[8]的研究可知,硫化态催化剂在实际反应过程中受催化剂活性位点影响很大,协作机制较为复杂,Pt作为贵金属加氢组分在接近常温常压的条件下就有很强的加氢活性,在工业上应用较广,作为吸附模型具有简单适用的特点,因此笔者选取Pt作为加氢活性组分。
根据前人的工作[9-13]可知,双功能催化剂中的加氢组分可能以团簇的形态存在。团簇是指粒径比纳米更小的原子聚集体[14],研究表明,金属纳米粒子催化剂能够表现出非常高的催化活性和选择性。Liu等[15]通过表征认为,分子筛孔道内稳定的Pt团簇尺寸范围为0.2~0.7 nm。然而由于目前对双功能催化剂中加氢组分与酸性组分间的具体结合位置、结合方式的认识仍很有限,尚未形成共识,因此笔者对加氢催化剂模型进行简化处理,构建Pt团簇作为加氢组分模型。团簇尺寸选取主要参考前人[16-20]对金属原子簇的研究,并充分考虑现有硬件条件下研究体系的总原子数对模拟耗时的影响,选取5原子的Pt团簇作为加氢催化剂的模型,标记为Pt5,优化结构如图2所示。
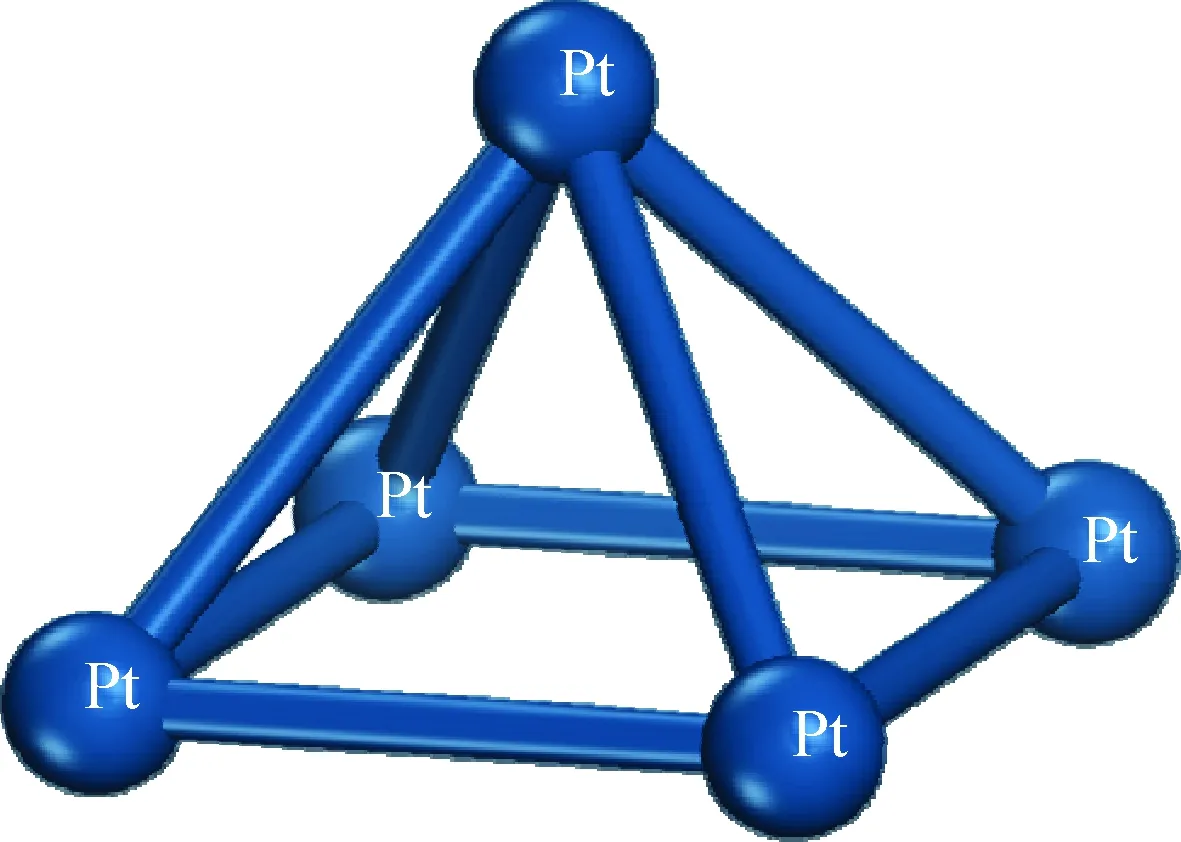
图2 Pt5团簇模型的构建Fig.2 Pt5 Cluster as hydrogenation catalyst model
经分析,本研究所构建的Pt5簇模型中各Pt-Pt间的距离均在0.27 nm左右,这一结果与前人[21-22]的研究结果基本一致,可作为本吸附研究的加氢中心。
2 结果与讨论
2.1 H2吸附与活化解离研究
为考察多环芳烃在Pt5团簇上的吸附,有必要确定H2在Pt5团簇上吸附并解离的可行性,将该单吸附体系记为H2-Pt5体系。笔者首先对1个H2分子在逐渐接近Pt5团簇过程中吸附热(Adsorption energy)随吸附距离(Adsorption distance between H2and Pt5cluster)的变化、及2个H原子间距离(H—H distance)随吸附距离的变化进行考察,从而初步确定H2在Pt5簇上发生吸附态转变并最终解离活化的过程。通过分别寻找H2与Pt5团簇的质心,并将两质心间的距离作为吸附距离,结果如图3 所示。
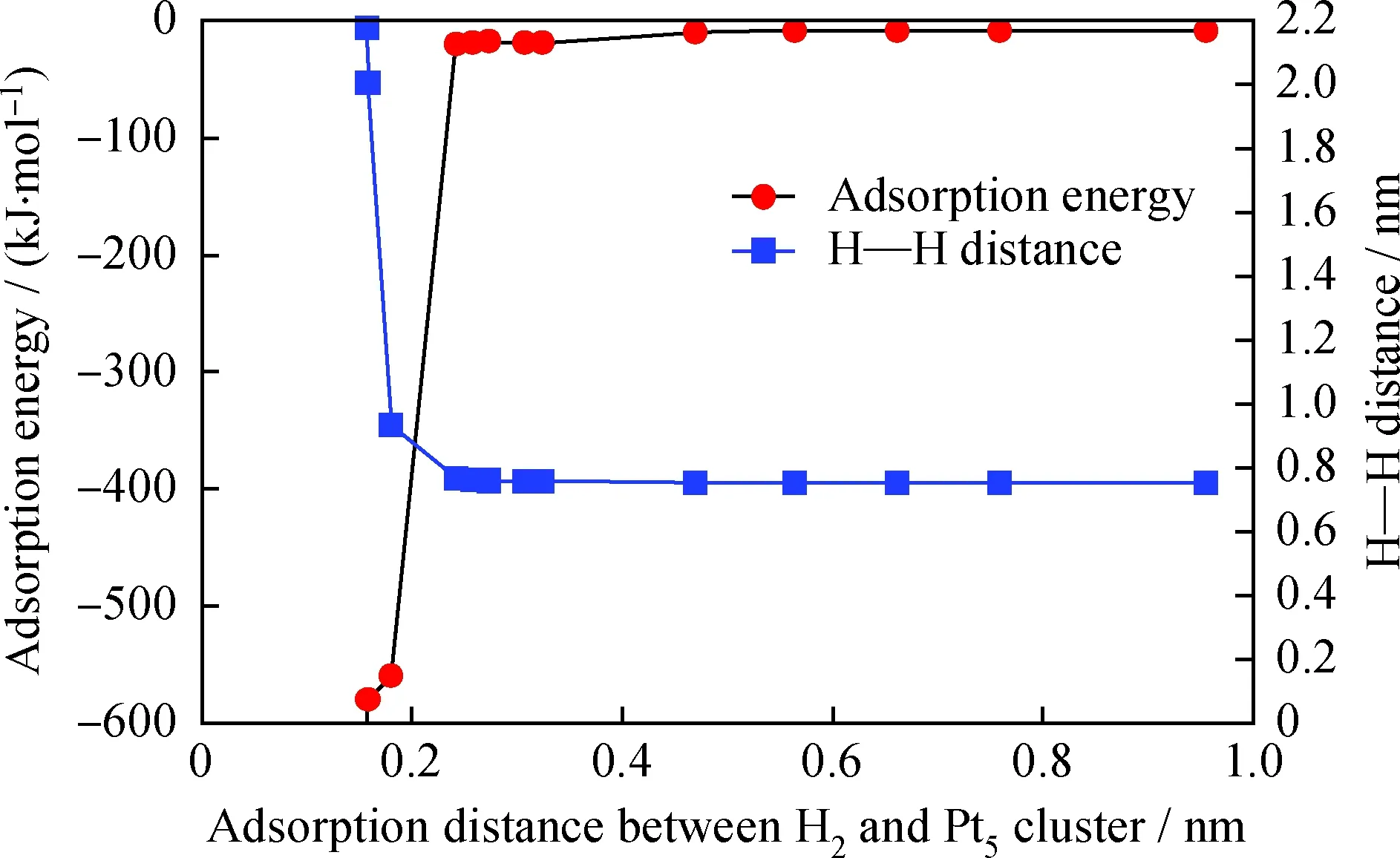
图3 H2-Pt5体系的吸附距离对H—H距离及吸附热的影响Fig.3 Effects of adsorption distance on H—H distance and adsorption heat
分析H2-Pt5体系的吸附热随吸附距离的变化可知,在吸附距离逐渐接近至0.24 nm时该体系释放的吸附热为18~20 kJ/mol,吸附作用较弱,符合物理吸附的吸附热特征(20~40 kJ/mol)[23],推测此时H2在Pt5上的吸附态为物理吸附;同时分析也发现,在这一过程中随H2-Pt5体系吸附距离的逐渐缩小吸附热有缓慢降低的趋势,可见H2有自发吸附到Pt5团簇表面的内在驱动力;在吸附距离从0.24 nm进一步接近至0.14 nm这一过程中,该体系释放的吸附热迅速增加,黑色吸附热曲线在此区间内迅速下降,这一过程释放的吸附热比典型物理吸附释放的吸附热高1个数量级,符合化学吸附的吸附热特征(40~400 kJ/mol),可以推知,在这一过程中发生了物理吸附至化学吸附的转变。当H原子继续接近Pt5团簇至小于0.14 nm时,该吸附体系的结构已极为不稳定,受模拟手段限制已无法得到收敛的吸附构象,故无小于0.14 nm的吸附热数据。
通过分析H—H原子间距离随吸附距离变化的曲线,结果也表明,当H2的质心与Pt5团簇的质心间距离大于0.24 nm时,H—H的原子间距离稳定在0.075 nm左右,此时H—H为稳定的H2分子,H2与Pt5团簇间的相互作用为弱范德华力,从而说明在本研究体系下当吸附距离大于0.24 nm时的吸附态确实为物理吸附;当距离接近至0.24 nm 时,H—H原子间距离迅速增加,大于0.075 nm,H原子与距离最近的Pt原子间距离约为0.158 nm, H—Pt原子间的距离小于H原子与Pt原子原子半径之和(H:0.25~0.38 nm、Pt:0.125~0.139 nm)[24],可以认为此时H原子与Pt原子间形成强的化学键力,进而验证了H2在距离Pt5团簇0.24 nm时发生了物理吸附至化学吸附的转变这一推测;H2与Pt5团簇的质心距离从0.24 nm进一步缩小至0.14 nm 的过程中,H—H间的距离继续迅速增大,可以认为此时氢键已发生彻底断裂,H2在Pt5团簇上发生了活化解离并形成2个活化H各自吸附于团簇中的2个Pt原子上,优化后得到的稳定的吸附构象如图4所示,笔者将这一化学吸附体系记为“2H-Pt5”。
此外,通过本部分的密度泛函研究还发现,H2在Pt5团簇上从物理吸附到化学吸附的转变过程仅需跨越1个极低的能垒,为10.79 kJ/mol,说明H2能够比较容易的在Pt5表面发生解离。上述结果与姚淑娟[21]对H2在Ptn团簇上解离的研究结果基本一致,也说明对于本研究构建的体系所使用的相关参数设置是合理的。
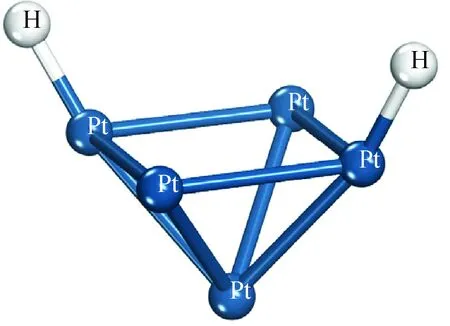
图4 H2在Pt5团簇的化学吸附构象Fig.4 Chemisorption conformation of 2H-Pt5 system
2.2 H2在Pt5团簇解离的电子机制
为更深入了解H2能够较容易地在Pt5团簇上解离活化的原因,笔者进一步从电子层面进行分析。
由于Pt原子最外层的价电子轨道排布为5d96s1,在5d轨道和6s轨道上各存在1个未成对电子处于能量较高的外层轨道上,H2具有空的反键轨道且能量较低。Pt的外层电子有可能从Pt原子上转移至H2反键轨道,进而弱化H—H键能并促进H2在Pt5表面解离并形成化学吸附,如图5 所示。
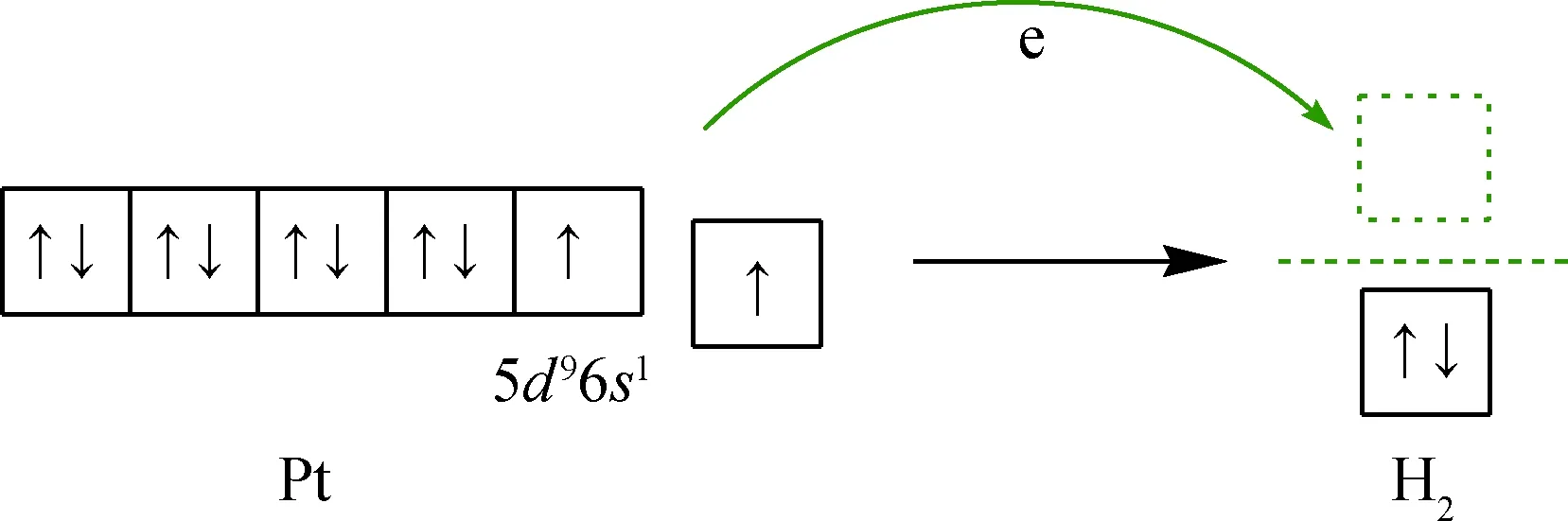
图5 H2与Pt5团簇相互作用的轨道分析Fig.5 Orbital analysis of H2 and Pt5 cluster interaction Green arrow: Indicating the transfer direction of electrons
为验证这一推测,进一步对物理吸附体系(H2-Pt5)与化学吸附体系(2H-Pt5)的电子密度分布进行分析与比较,电子云分布如图6所示。
电子密度分布表明,物理吸附体系中H2分子周围的电子云密度低于Pt5团簇,Pt5团簇上的电子密度相对较高且主要集中在2个3配位的Pt原子上。发生化学吸附后,Pt5团簇上的电子密度相对于H2-Pt5体系整体略有降低,位于团簇两端的2个三配位Pt原子以及与活化H成键的Pt原子附近电子云密度降低,未占据的四配位Pt原子以及活化H上的电子密度显著增加,可见在体系从物理吸附转变为化学吸附过程中,Pt5团簇中朝向活化H一面的几个Pt原子上的电子迁出并向活化H转移。为更明确电子的迁移方向,继续对H2-Pt5物理吸附体系和2H-Pt5化学吸附体系中的吸附质(Adsorbate)、吸附剂(Adsorbent)分别进行电荷分布分析,将Pt5团簇整体视为吸附剂,H2和解离生成的2个活化H分别视为2种吸附体系中的吸附质,在不同吸附态下各自所带的总电荷分析结果如表2所示。
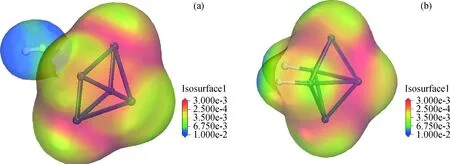
图6 H2吸附于Pt5团簇上的电子密度分析Fig.6 Electron density analysis of H2 adsorbed on Pt5 cluster(a) H2-Pt5 adsorption system; (b) 2H-Pt5 adsorption system Isosuface: Indicating the electron density distribution

表2 H2在Pt5团簇上生成物理、化学单吸附体系过程中 吸附质、吸附剂所带的单位电荷分析Table 2 Charge distribution of the single molecular adsorption systems
由电荷分析可知,物理吸附态下,吸附质、吸附剂各自呈电中性,所带电荷为0;在活化解离过程中总共有0.143个单位的电子从Pt5团簇转移至活化H,电荷分析也发现,Pt5团簇上带正电荷最多的原子主要为邻近H原子的几个Pt原子,说明填入H原子反键轨道的电子主要来自邻近H原子的几个Pt原子的外层轨道,充分证明了之前从电子轨道角度进行的推测是正确的。
综合上述分析,本研究体系中当H2接近Pt5团簇至化学吸附的临界距离时,邻近的Pt原子的外层电子迁入H2空的反键轨道, H—H间的作用减弱,H—H键能降低,与此同时,H-Pt间作用强度增强,从而促进H2在Pt团簇表面解离,这是H2能够较容易地在Pt5团簇上解离的内在电子机制。
2.3 壬基蒽吸附研究
为确定壬基蒽在Pt5团簇上吸附的可行性,采用相同的研究方法首先考察了9-壬基蒽在纯Pt5团簇上单吸附(9-Nonyl anthracene-Pt5)以及在解离有活化H的Pt5团簇上共吸附(9-Nonyl anthracene-2H-Pt5)的吸附热,并与化学吸附态的2H-Pt5单吸附体系进行比较,结果如表3所示。

表3 壬基蒽单独、共吸附时的吸附热 Table 3 Adsorption heat of 9-nonyl anthracene on Pt5 and 2H-Pt5 system
由表3可知,H2在Pt5团簇表面吸附作用最强,9-壬基蒽在有活化H的Pt5团簇表面的吸附强度次之。通过比较可知,H2能够优先在Pt5团簇上发生化学吸附,从而为9-壬基蒽的催化加氢创造了条件;9-壬基蒽将优先吸附于解离有活化H的Pt5团簇表面,为后续催化加氢创造了可能。优化9-壬基蒽在单独吸附、共吸附时的稳定吸附构象,结果如图7所示。
2.4 壬基蒽共吸附体系的前线轨道分析
利用前线轨道理论[25]对9-壬基蒽与活化H结合并加氢的可行性及电子作用机制进行深入研究。由于壬基蒽在团簇上的吸附距离大于活化H与团簇的吸附距离,可将2H-Pt5体系与壬基蒽视为2种反应物分别进行前线轨道分析。首先分析9-壬基蒽的前线轨道分布。图8为9-壬基蒽前线轨道分布。
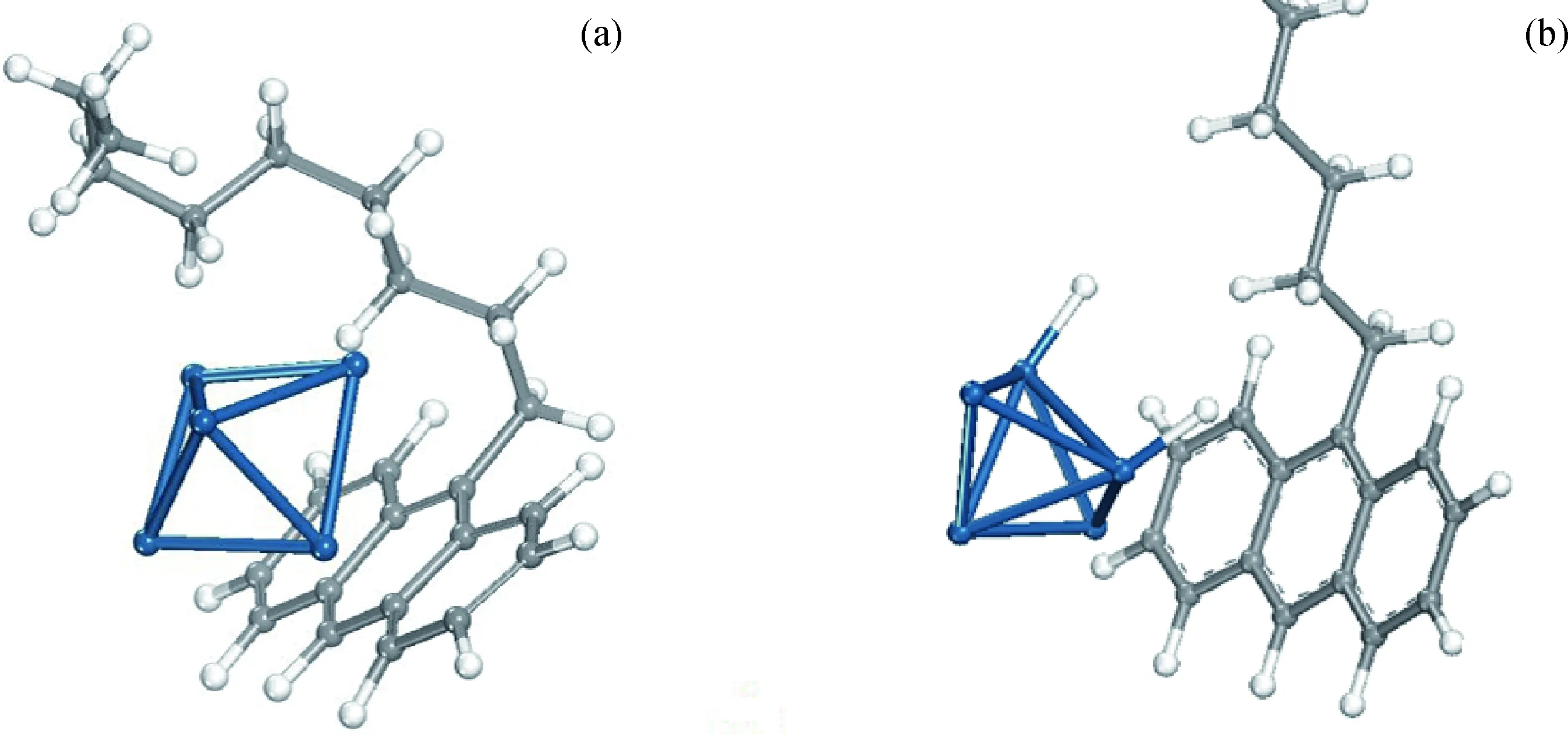
图7 9-壬基蒽单独吸附、共吸附时的吸附构象Fig.7 Adsorption configuration of 9-nonyl anthracene adsorption systems(a) 9- Nonyl anthracene-Pt5; (b) 9- Nonyl anthracene-2H-Pt5 Blue: Pt atoms; Grey: Carbon atoms; White: Hydrogen atoms
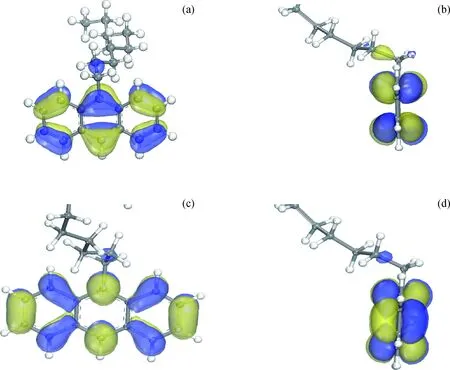
图8 9-壬基蒽前线轨道分布Fig.8 Frontier orbital distribution of 9-nonyl anthracene(a) Vertical view of 9-nonyl anthracene’s HOMO orbital; (b) Side view of 9-nonyl anthracene’s HOMO orbital; (c) Vertical view of 9-nonyl anthracene’s LUMO orbital; (d) Side view of 9-nonyl anthracene’s LUMO orbital
由图8可知,壬基蒽的HOMO轨道和LUMO轨道均集中分布于芳香环上,由此可知芳香环是9-壬基蒽发生加氢反应的主要位置。为进一步比较该共吸附体系前线轨道的能量分布,将9-壬基蒽视为吸附质、将化学吸附体系的2H-Pt5视为吸附剂,吸附剂与吸附质的前线轨道能量分析结果如表4所示。
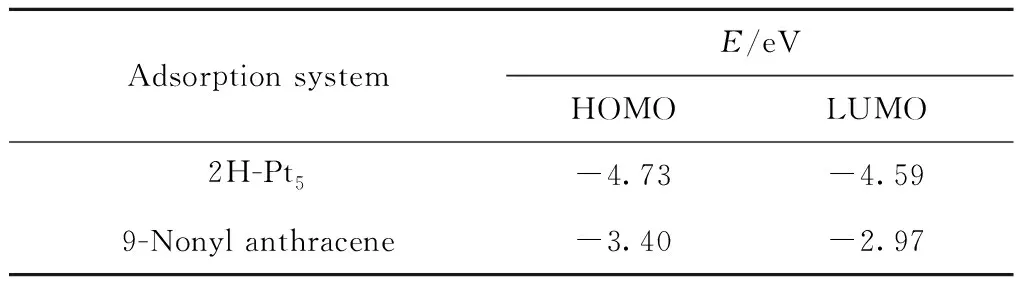
表4 9-壬基蒽与2H-Pt5体系的前线轨道能量Table 4 Frontier orbital energies of 9-nonyl anthracene and 2H-Pt5
其中2H-Pt5的HOMO轨道与9-壬基蒽的LUMO轨道的能级差为1.76 eV,2H-Pt5体系的LUMO轨道与9-壬基蒽的HOMO轨道的能级差为1.19 eV,可见2H-Pt5的LUMO轨道与9-壬基蒽的HOMO轨道能量更接近。进一步分析2H-Pt5体系的LUMO轨道与9-壬基蒽的HOMO轨道的空间分布,如图9所示,可以看出轨道的对称性也是匹配的。
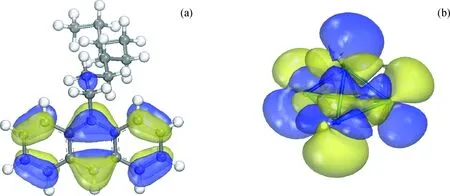
图9 前线轨道的空间分布Fig.9 Steric distribution of frontier orbitals(a) HOMO orbital distribution of 9-nonyl anthracene; (b) LUMO orbital distribution of 2H-Pt5
进一步对单吸附、共吸附体系下吸附质、吸附剂各自所带的电荷进行分析与比较,对2H-Pt5体系来说可将解离的活化H视为吸附质,将Pt5簇视为吸附剂。对9-nonyl anthracene-Pt5体系来说可将9-壬基蒽视为吸附质,Pt5簇视为吸附剂。对9-nonyl anthracene-2H-Pt5体系来说,可将9-壬基蒽和解离的活化H视为吸附质,Pt5簇视为吸附剂。分析结果如表5所示。

表5 几种吸附体系中吸附质、吸附剂所带电荷分析Table 5 Charge distribution of adsorbates and adsorbents
分析电荷的分布情况显示,2种单吸附体系中的吸附质(活化H、9-壬基蒽)皆倾向从吸附剂(Pt5)夺取电子,从而令吸附质带负电荷,吸附剂带正电荷,其中活化H夺取电子的能力强于9-壬基蒽。当2种吸附质共存时,9-壬基蒽带正电荷,2H-Pt5带负电荷,其中由于2H-Pt5体系中的活化H夺电子能力较强,所带的负电荷多于Pt5,而9-壬基蒽夺电子能力相对较弱,在共吸附体系中失去电子,2H-Pt5成为夺电子体。可见在单独吸附时,由于Pt原子对外层电子的束缚能力低于9-壬基蒽,也弱于活化H,容易供出电子给9-壬基蒽与活化H,这一电子传递方向与前述推测的前线轨道的作用方式是相一致的。
综合上述分析,9-壬基蒽的HOMO轨道与2H-Pt5体系的LUMO轨道能量接近,轨道对称性匹配,电子转移方向与发生作用的前线轨道相一致,由此可知,9-壬基蒽在Pt5团簇上的催化加氢是通过9-壬基蒽的HOMO轨道与2H-Pt5的LUMO轨道相互作用进行的。
2.5 共吸附体系电子作用机制的提出
结合对吸附热、电子分布等方面分析,笔者提出了在9-nonyl anthracene-2H-Pt5共吸附体系中,9-壬基蒽与吸附于Pt团簇上的活化H相互作用的内在电子机制,该电子传递机制可绘制为如图10所示的电子传递环路。
当9-壬基蒽通过π轨道与2H-Pt5体系的d轨道发生相互作用时,由于9-壬基蒽对电子的束缚能力弱于2H-Pt5体系,电子从HOMO轨道迁入2H-Pt5体系的LUMO轨道。这一电子交换作用改变了2H-Pt5体系中的电荷分布情况,一方面削弱了Pt5团簇与活化氢之间的相互作用,另一方面由于9-壬基蒽芳香环上电子的转出使芳香碳呈现出强的缺电子活性。在2种作用下活化H得以从Pt5团簇表面脱附并与9-壬基蒽芳香环上的不饱和芳香碳结合,从而实现对9-壬基蒽芳环的加氢。
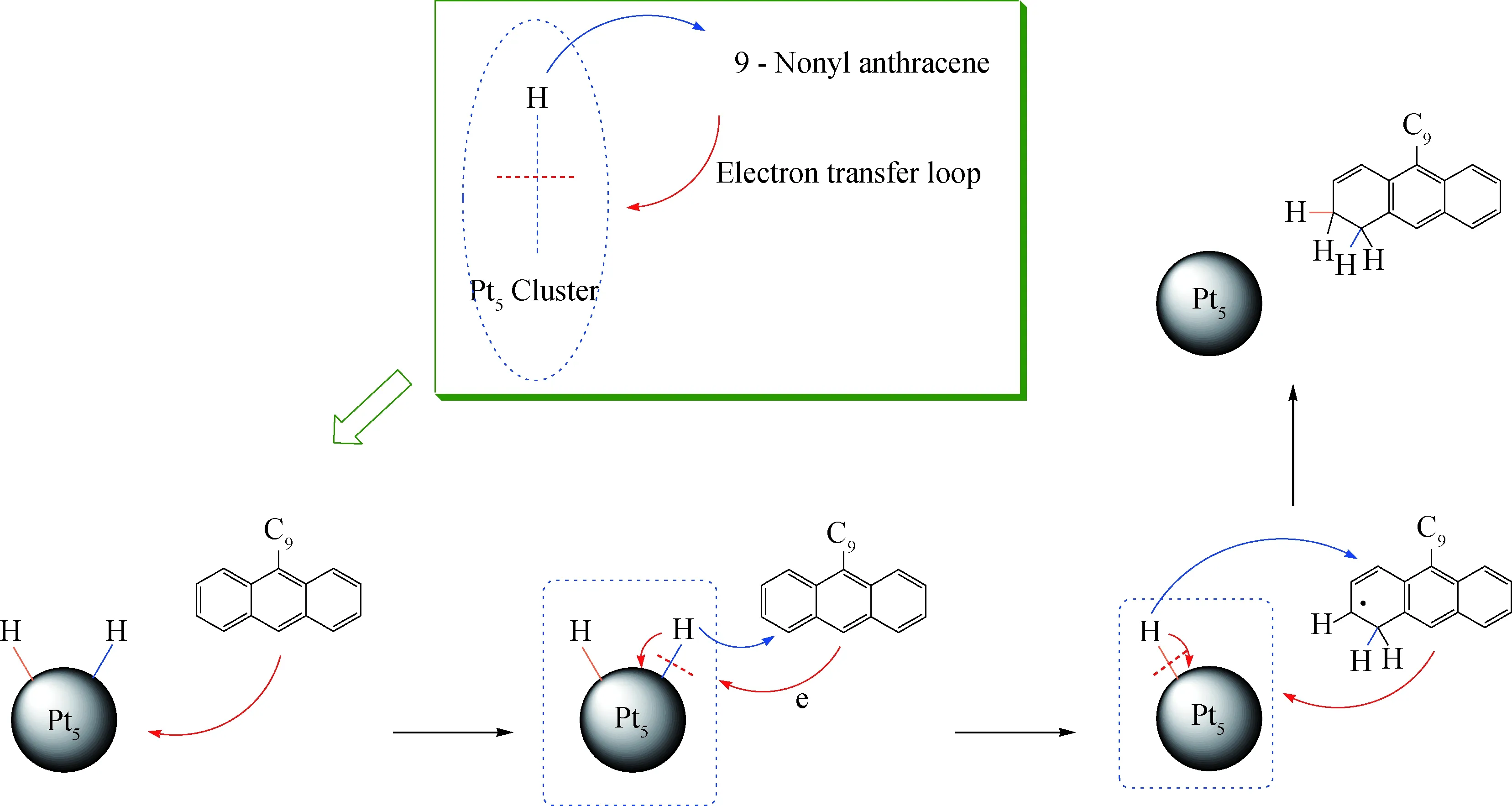
图10 共吸附体系的电子作用机制示意图Fig.10 Orbital interaction mechanism illustration of the co-adsorption systems
3 结 论
通过H2及9-壬基蒽在Pt团簇上的一系列单吸附、共吸附的初步研究,进一步明确了H2及多环芳烃在过渡金属催化剂上的吸附加氢的内在机制,能够对催化剂的设计开发有一定理论指导意义。本研究体系主要得出以下结论。
(1)通过考察H2在距离Pt团簇不同距离时的吸附热,可知H2能够自发物理吸附到Pt5团簇表面。
(2)H2能够较容易的在Pt5表面解离活化。其原因是由于Pt原子外层电子排布特性,能够将外层电子填入H2的反键轨道,从而弱化H—H间相互作用,促进H2在Pt团簇表面的活化解离。
(3)与裸环结构的稠环芳烃在过渡金属表面的吸附类似,带有长取代侧链的9-壬基蒽同样通过分布于芳环上的离域大π键吸附于解离有活化氢的Pt5团簇表面,从而为在Pt5团簇表面的催化加氢创造了可能。
(4)9-壬基蒽通过HOMO轨道与解离有活化H的Pt团簇的LUMO轨道发生作用,电子首先从9-壬基蒽的HOMO轨道迁入解离有活化H的Pt团簇的LUMO轨道,在电子的迁移作用下,活化H与Pt5团簇间的作用强度减弱,同时壬基蒽的缺电子活性增加,从而具备了在解离有活化H的Pt5团簇表面催化加氢的条件。
猜你喜欢
化工管理(2022年13期)2022-12-02
科学家(2021年24期)2021-04-25
能源工程(2021年1期)2021-04-13
湿法冶金(2020年6期)2020-12-21
合成化学(2015年9期)2016-01-17
橡胶工业(2015年9期)2015-08-29
橡胶工业(2015年6期)2015-07-29
橡胶工业(2015年4期)2015-07-29
浙江理工大学学报(自然科学版)(2015年5期)2015-03-01
中国洗涤用品工业(2015年9期)2015-02-28
