
顶空气相色谱-质谱法测定依度沙班原料药中遗传毒性物质
2019-01-16 08:41徐艳梅裴丽娟杜高锋宋更申
医药导报 2019年1期
徐艳梅,裴丽娟,杜高锋,宋更申
(1.河北省药品检验研究院,石家庄 050011;2.河北医科大学第一医院药剂科, 石家庄 050031)
依度沙班(edoxyaban)是一种新型Xa因子抑制药[1-2],具有起效快、抗凝效果可逆、具有剂量依赖的效果等特点。2015年以来,美国食品药品管理局(FDA)、欧洲药品管理局(EMA)和瑞士药品管理局相继批准依度沙班用于非瓣膜性心房颤动患者脑卒中及系统性栓塞、急性静脉栓塞患者深静脉血栓和肺栓塞的预防及治疗[3]。依度沙班原料药在合成工艺中有一步甲磺酰化的过程,其中甲磺酰氯易与醇类反应产生甲磺酸酯[4]。依度沙班合成工艺中将甲醇和乙醇作为反应溶剂,因此依度沙班原料药中可能存在甲磺酸甲酯(methyl methanesulfonate)、甲磺酸乙酯(ethyl methanesulfonate)及甲磺酸异丙酯(isopropylmethanesulfonate)3种杂质。甲磺酸酯类物质是一种潜在性基因毒性杂质[5-6],这些杂质的DNA烷基化作用会产生诱变效应、致癌效应和致畸效应[7],严重威胁人类健康,因此需要严格控制药品中甲磺酸酯的限度。根据EMEA发布的《遗传毒性杂质限度指导原则》相关规定,按照毒理学担忧阈值(TTC)作为评价大部分遗传毒性杂质的阈值,则遗传毒性杂质摄入量最大限值为1.5 μg·d-1[8-9]。依度沙班作为抗凝药物使用时,每日通常使用剂量为30 mg,按此计算含甲磺酸甲酯、甲磺酸乙酯、甲磺酸异丙酯不得过50 μg·g-1。近年来,气相色谱(GC)法、气相色谱-质谱联用(GC-MS)法和衍生化-气相色谱-质谱联用(HS-GC-MS)法已被广泛应用于甲磺酸酯类基因毒性杂质检测[10-11]。笔者参考《欧洲药典》(EP)8.0 版中关于甲磺酸盐中甲磺酸甲酯、甲磺酸乙酯、甲磺酸异丙酯的测定方法,采用HS-GC-MS法对甲磺酸依度沙班原料药中上述3 种遗传毒性杂质进行测定。
1 仪器与试药
1.1仪器 GC-MS 2010 Plus色谱仪(日本Shimadzu公司);DU305型电子天平(瑞士Mettler-Toledo公司,感量:0.01 mg)。
1.2试药 依度沙班原料药(河北志恒医药科技有限公司,批号:160031,160032,160033);甲磺酸甲酯对照品(JSK SCIENTIFIC LTD,批号:LU80L27);甲磺酸乙酯对照品(Atta Aesar,批号:10169291);甲磺酸异丙酯对照品(ACROS,批号:A0314222);甲磺酸丁酯(南京强山化工有限公司,批号:20130027);乙腈和水均为色谱纯,无水碘化钠和硫代硫酸钠均为分析纯。
2 方法与结果
2.1GC-MS条件 色谱柱为DB-WAX毛细管柱(30 m×0.25 mm,0.25 μm);检测器为MS检测器;进样口温度为110 ℃;采用程序升温,起始温度为40 ℃,维持3 min,以20 ℃·min-1的速率升温至150 ℃,维持2 min,然后以20 ℃·min-1的速率升温至230 ℃,维持10 min;进样方式为分流进样,分流比20:1;载气为高纯氦气;柱流量为0.6 mL·min-1;顶空进样,炉温60 ℃;平衡时间30 min;进样体积1 mL,进样时间1 min;离子源温度200 ℃;接口温度150 ℃;扫描方式为选择离子扫描(SIM);电子能量为70 eV。各待测成分的定性、定量信息见表1,质谱图见图1。
表1待测成分的定性、定量信息
Tab.1Qualitativeandquantativedataoftheingredientundertest
2.2溶液配制
2.2.1衍生化试剂 精密称取无水碘化钠60.0 g和硫代硫酸钠30 mg置50 mL量瓶中,加水溶解并稀释至刻度,摇匀,即得。
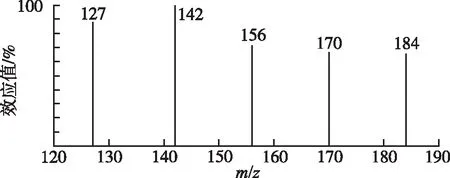
图1碘代甲烷、碘代乙烷、碘代异丙烷和碘代丁烷的质谱图
Fig.1Massspetrumofmethyliodide,ethyliodide,iso-propyliodideandiodo-2-methylpropane
2.2.2内标溶液 精密称取甲磺酸丁酯25 mg,置25 mL量瓶中,加稀释液(水-乙腈20:80)溶解并稀释至刻度,摇匀,精密量取该溶液50 μL,置于10 mL量瓶中,加稀释液稀释至刻度,摇匀,另精密量取该溶液2 mL,至50 mL量瓶中,加稀释液稀释并定容至刻度,摇匀,作为内标溶液。
2.2.3空白溶液 精密量取水-乙腈(20:80)0.5 mL与内标溶液0.5 mL,置20 mL顶空瓶中,立即密封,摇匀,作为空白溶液。
2.2.4对照品溶液 精密称取甲磺酸甲酯、甲磺酸乙酯、甲磺酸异丙酯各约50 mg,分别加甲苯溶解并稀释至10 mL,摇匀;精密量取50 μL,置同一25 mL量瓶中,加内标溶液稀释并定容,摇匀,作为对照品贮备液。精密量取对照品贮备液150 μL,置10 mL量瓶中,加内标溶液稀释定容,即得。分别精密量取该溶液0.5 mL及衍生化试剂0.5 mL,置20 mL顶空瓶中,立即密封,摇匀,作为对照品溶液。
2.2.5供试品溶液 精密称取样品30 mg,置20 mL顶空瓶中,加0.5 mL内标溶液及0.5 mL衍生化试剂,立即压盖密封,超声溶解,作为供试品溶液。
2.3专属性考察 取空白溶液、甲磺酸甲酯、甲磺酸乙酯、甲磺酸异丙酯对照品溶液和供试品溶液各1 mL,按“2.1”项色谱条件,记录色谱图。内标衍生物碘代丁烷保留时间为5.52 min,甲磺酸甲酯、甲磺酸乙酯和甲磺酸异丙酯的衍生物碘代甲烷、碘代乙烷和碘代异丙烷的保留时间分别为2.87,3.52,3.81 min,各峰之间的分离度均>2.0,符合检测要求。
2.4检测限和定量限考察 取对照品溶液,加内标液逐级稀释成不同质量浓度,分别精密量取该溶液0.5 mL及衍生化试剂0.5 mL,置20 mL顶空瓶中,摇匀,立即密封。按“2.1”项色谱条件分别进样,测定各杂质的检测限(S/N=3)和定量限(S/N=10)。结果,甲磺酸甲酯、甲磺酸乙酯和甲磺酸异丙酯检测限分别为5,5和10 ng·mL-1,定量限为15,15和30 ng·mL-1。
2.5线性关系考察 精密量取对照品贮备溶液0.05,0.1,0.5,1.0,3.0 mL,置10 mL量瓶中,加内标溶液稀释定容,制得3种成分浓度分别约0.05,0.1,0.5,1.0,3.0 μg·mL-1,分别精密量取该溶液0.5 mL及衍生化试剂0.5 mL,置20 mL顶空瓶中,立即密封,摇匀,作为线性工作溶液。按“2.1”项下色谱条件分别进样,记录色谱图。以对照品溶液的质量分数为横坐标,主成分与内标峰面积比值为纵坐标,绘制标准曲线,3种杂质的线性结果见表2。实验结果表明,甲磺酸甲酯、甲磺酸乙酯和甲磺酸异丙酯均在0.05~3.0 μg·mL-1浓度范围内线性关系良好。
表23种成分的线性结果
Tab.2Linearityofthreekindsofcomponents
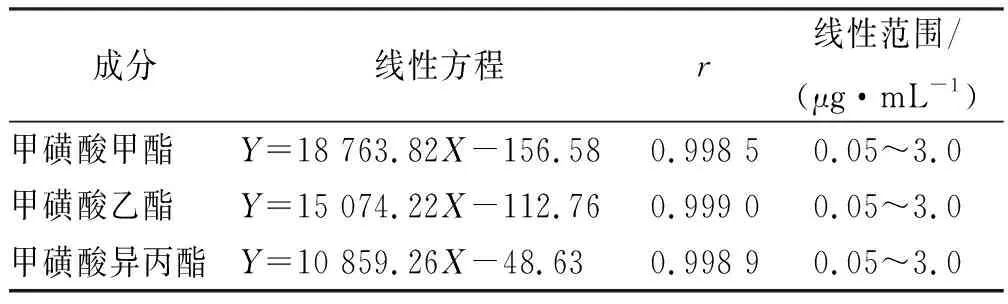
成分线性方程r线性范围/(μg·mL-1)甲磺酸甲酯Y=18 763.82X-156.580.998 50.05~3.0甲磺酸乙酯Y=15 074.22X-112.760.999 00.05~3.0甲磺酸异丙酯Y=10 859.26X-48.630.998 90.05~3.0
2.6精密度实验 取“2.5”项下线性工作溶液(3),按“2.1”项色谱条件连续进样6次,记录色谱图。经计算得A甲磺酸甲酯/A内标、A甲磺酸乙酯/A内标和A甲磺酸异丙酯/A内标的RSD分别为2.4%,3.0%,2.9%(n=6),表明仪器精密度良好。
2.7重复性实验 精密称取样品(批号:160031)30 mg,置20 mL顶空瓶中,加入0.15 μg·mL-1标准溶液0.5 mL及衍生化试剂0.5 mL,立即压盖密封,超声溶解,按“2.1”项色谱条件分别进样,记录色谱图。测定加标样品中甲磺酸甲酯、甲磺酸乙酯和甲磺酸异丙酯的含量,平行测定6次。结果表明该方法重复性良好,满足测定要求。
2.8加样回收率实验 精密称取样品(批号:160031)30 mg,共9份,分别置于20 mL顶空瓶中,精密加入对照品贮备溶液(10 μg·mL-1)2.25, 1.50和0.75 mL,每种浓度平行制备3份进行测定,分别加入衍生化试剂0.5 mL,立即压盖密封,超声溶解,按“2.1”项下色谱条件分别进样,记录色谱图。根据内标法计算甲磺酸甲酯、甲磺酸乙酯和甲磺酸异丙酯的平均回收率(n=9)分别为96.4%,96.1%和96.5%。结果见表3。
2.9样品含量测定 按“2.1”项色谱条件和“2.2”项对照品溶液和供试品溶液的制备方法操作,测定3批原料药中3种遗传毒性物质,以内标法计算甲磺酸甲酯、甲磺酸乙酯和甲磺酸异丙酯的含量,测定结果见表4。
表33种成分的回收率结果
Tab.3Recoveryresultsofthreekindsofcomponents
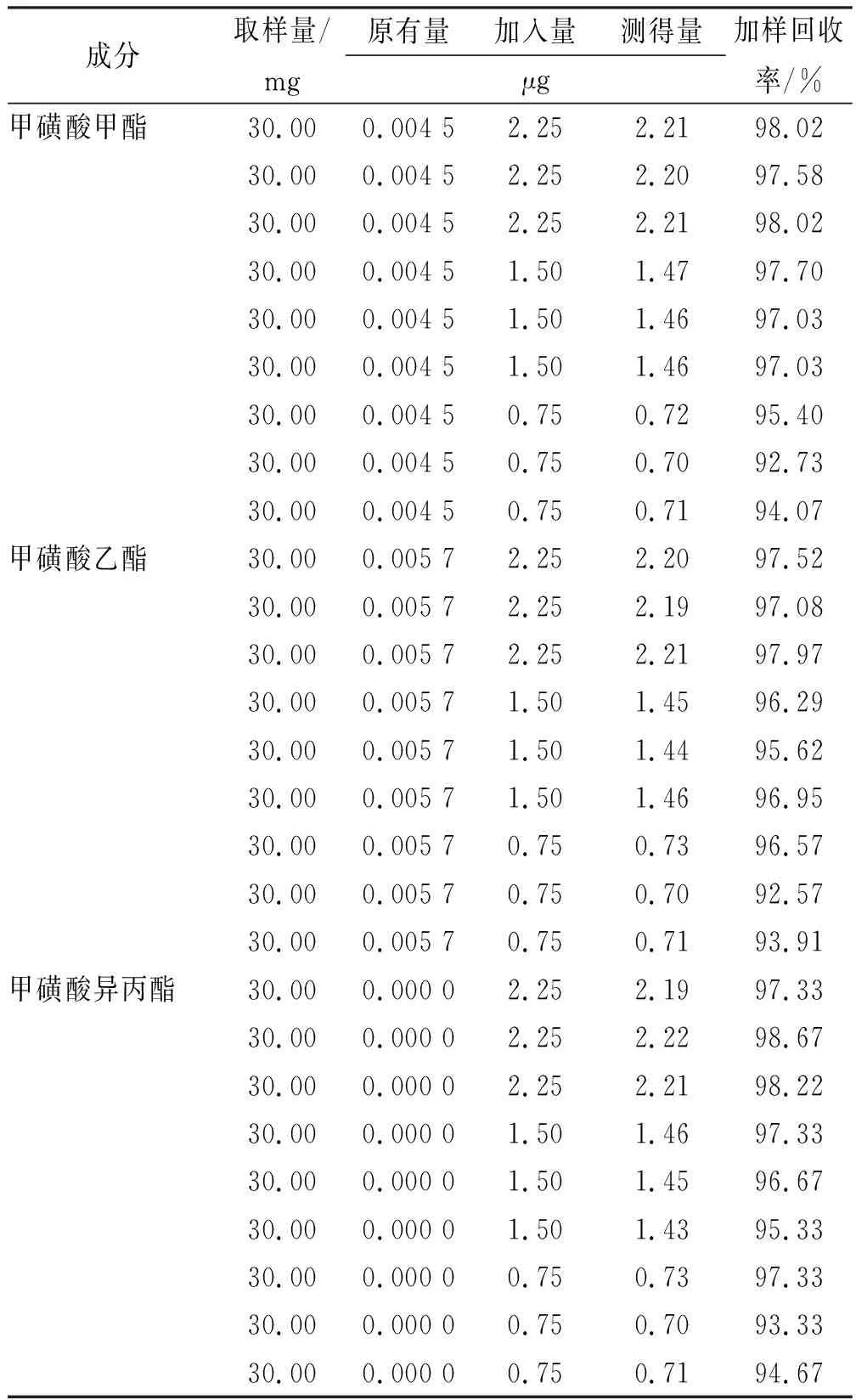
成分取样量/mg原有量加入量测得量μg加样回收率/%甲磺酸甲酯30.000.004 52.252.2198.0230.000.004 52.252.2097.5830.000.004 52.252.2198.0230.000.004 51.501.4797.7030.000.004 51.501.4697.0330.000.004 51.501.4697.0330.000.004 50.750.7295.4030.000.004 50.750.7092.7330.000.004 50.750.7194.07甲磺酸乙酯30.000.005 72.252.2097.5230.000.005 72.252.1997.0830.000.005 72.252.2197.9730.000.005 71.501.4596.2930.000.005 71.501.4495.6230.000.005 71.501.4696.9530.000.005 70.750.7396.5730.000.005 70.750.7092.5730.000.005 70.750.7193.91甲磺酸异丙酯30.000.000 02.252.1997.3330.000.000 02.252.2298.6730.000.000 02.252.2198.2230.000.000 01.501.4697.3330.000.000 01.501.4596.6730.000.000 01.501.4395.3330.000.000 00.750.7397.3330.000.000 00.750.7093.3330.000.000 00.750.7194.67
表4样品中3种遗传毒性物质含量测定结果
Tab.4Contentdeterminationonthreekindsofgenotoxicimpuritiesinsamplesμg·g-1
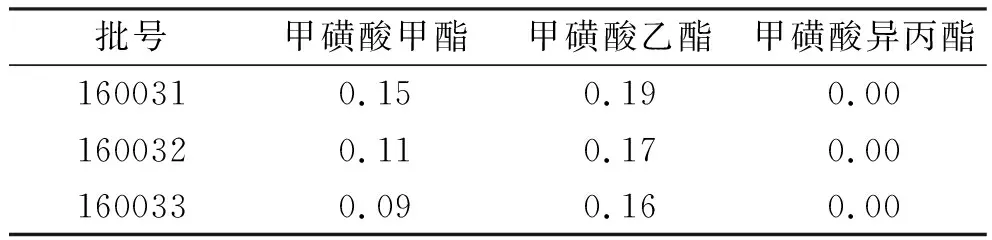
批号甲磺酸甲酯甲磺酸乙酯甲磺酸异丙酯1600310.150.190.001600320.110.170.001600330.090.160.00
3 讨论
本研究采用内标法进行定量分析,选取待测物的同系物甲磺酸丁酯作为内标物,其衍生化产物为碘代丁烷,与待测物质完全分离,且灵敏度较高。因此,甲磺酸丁酯可作为这3种遗传毒性杂质含量测定的内标物。
因本品理化性质原因,溶解性较差,在溶剂筛选过程中分别考察了甲醇、异丙醇、二氯甲烷和乙腈等溶剂。实验结果表明,选用混合溶剂水-乙腈(20:80)效果最佳。
实验中考察了不同极性的色谱柱,结果表明,3种待测组分和内标物在DB-WAX毛细管柱(30 m×0.25 mm,0.25 μm)上的峰形和分离度最佳。
实验采用DB-WAX毛细管柱内径为0.25 mm,采用常用流速1.0 mL·min-1不能使3种待测组分和内标物达到分离度的要求,故考察了1.0,0.8和0.6 mL·min-13种不同流速对分离度的影响,结果表明,流速为0.6 mL·min-1时,分离效果最好。
色谱分离时,不仅载气流速影响分离度,而且升温程序的不同也会导致色谱峰分离度的变化。由于3种待测组分和内标物的沸点接近,为了使之达到很好的分离,需采用较低的起始温度进行实验,最终选择40 ℃作为起始温度,程序升温进行实验。
顶空条件的优化主要考察了两个方面,包括炉温和平衡时间的选择。实验考察了不同炉温对实验结果的影响,结果表明,炉温为60 ℃时,能够使3种待测组分和内标物充分气化。不仅炉温会影响气化程度,平衡时间也会影响气化程度,不同的平衡时间直接影响顶空进样的浓度,故考察了平衡时间的影响,最终选择平衡时间为30 min。
本研究采用HS-GC-MS方法对依度沙班合成过程中可能产生的基因毒性物质甲磺酸甲酯、甲磺酸乙酯和甲磺酸异丙酯进行含量测定,对于确保临床用药的安全性和有效性具有重要的意义。
猜你喜欢
酿酒科技(2022年8期)2022-08-20
中国典型病例大全(2022年9期)2022-04-19
云南化工(2020年11期)2021-01-14
中成药(2018年5期)2018-06-06
中国粮油学报(2016年5期)2016-01-23
中国卫生标准管理(2015年14期)2016-01-15
烟草科技(2015年8期)2015-12-20
中南民族大学学报(自然科学版)(2015年2期)2015-12-16
中国当代医药(2015年10期)2015-03-01
中国当代医药(2015年9期)2015-03-01
