
新精神活性物质Dibutylone的鉴定
2019-02-10 11:15王跨陡曹芳琦江雪陈红袁晓亮陈永生胡钧健
法医学杂志 2019年6期
王跨陡,曹芳琦,江雪,陈红,袁晓亮,陈永生,胡钧健
(1.上海市公安局物证鉴定中心 上海市现场物证重点实验室,上海 200083;2.上海市刑事科学技术研究院 上海市现场物证重点实验室,上海 200083)
Dibutylone(bk-DMBDB)中文名为1-[3,4-(亚甲二氧基)苯基]-2-二甲氨基-1-丁酮,是一种合成卡西酮类新精神活性物质,其分子式是C13H17NO3,相对分子质量为235.28,分子结构见图1。Dibutylone与浴盐的主要成分亚甲基二氧吡咯戊酮(methylenedioxypy⁃rovalerone,MDPV)、3,4-亚甲二氧基甲卡西酮(3,4-methylenedioxy-N-methylcathinone,methylone,bk-MDMA)、2-甲氨基-1-[3,4-(亚甲二氧基)苯基]-1-丁酮[1-(3,4-methylenedioxyphenyl)-2-methylamino⁃butan-1-one,butylone,bk-MBDB]具有相同的基本结构(图1),都属于亚甲二氧基取代的苯乙胺类β酮化合物。20世纪60年代首次在专利报道中出现Dibu⁃tylone,近年来Dibutylone却作为一种新型策划药在欧美等地流行[1]。2010年,芬兰向欧洲毒品与毒瘾监测中心(European Monitoring Centre for Drugs and Drug Addiction,EMCDDA)及欧洲刑警组织首次正式通报了Dibutylone[2]。此后,在瑞典STRIDA项目(一个研究新型滥用药物在瑞典造成健康危害的项目)收集的粉末样品中检出了Dibutylone成分[3]。2016年,美国国家法医实验室信息系统(National Fo⁃rensic Laboratory Information System,NFLIS)年报显示,在全年346 681例苯乙胺类毒品的报告中,Dibu⁃tylone有2 000例,占总数的0.58%,仅次于甲基苯丙胺(90.82%)、苯丙胺(3.62%)和3,4-亚甲二氧基甲基苯丙胺(methylene dioxymethamphetamine,MDMA,1.65%),排名第4[4]。2017年,尽管Dibutylone的报告案例数和所占比例有所下降,但仍在苯乙胺类β酮毒品中排名第2,仅次于N-ethylpentylone[5]。目前,Dibutylone作为1-[3,4-(亚甲二氧基)苯基]-2-甲氨基-1-戊酮[1-(3,4-methylenedioxyphenyl)-2-(me⁃thylamino)pentan-1-one,pentylone]的位置异构体在美国被列为管制药物法附表一级(ScheduleⅠ)管制物质。我国在2018年9月1日已将其列入了《非药用类麻醉药品和精神药品管制品种增补目录》中予以管控。为了逃避法律管控,不法分子对该类物质的分子结构进行微调,因此出现了越来越多具有相同基本结构和类似药效的结构类似物,也使得该类物质在全球范围的流行趋势不断更新变化。目前,我国已列管了12种亚甲二氧基取代的苯乙胺类β酮物质。
目前国内外关于Dibutylone检验方法的文献报道较少。GWAK等[6]采用离子迁移谱和实时直接分析-四极杆飞行时间质谱建立了对包含Dibutylone在内的35种新精神活性物质进行快速筛查的方法。LEHMANN等[7]开发了在线固相萃取结合液相色谱-串联质谱(liquid chromatography-tandem mass spec⁃trometry,LC-MS/MS)的方法快速分析血清中包括Dibutylone在内的74种新精神活性物质,并进行了方法学验证。KROTULSKI等[1]采用LC-MS/MS法对人血液、尿液、玻璃体液、唾液和肝组织中的Dibutylone进行了定性定量分析,并应用体外人肝微粒体孵育体系对其代谢情况进行研究。本研究采用气相色谱-质谱(gas chromatography-mass spectrometry,GC-MS)、高分辨质谱的分析方法对未知样品进行检测,筛查出Dibutylone,经硅胶柱层析纯化后又用核磁共振氢谱(1H nuclear magnetic resonance spectroscopy,1H NMR)进一步确证其结构。
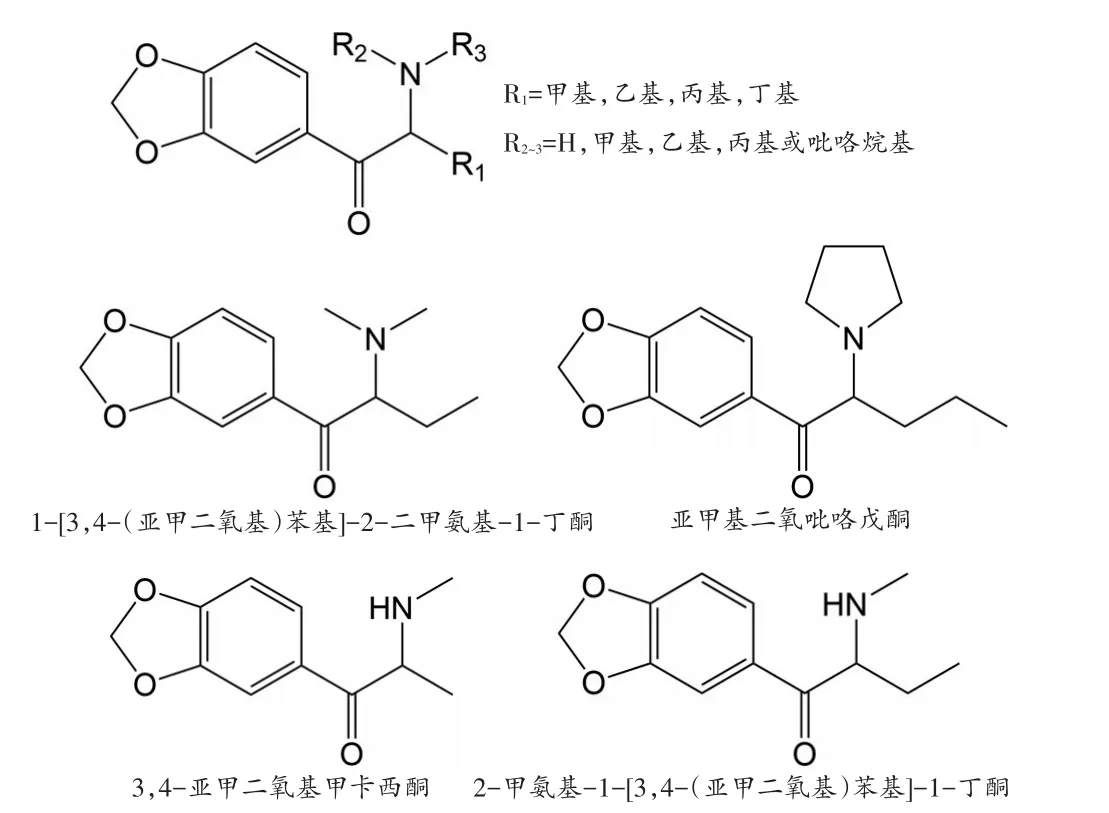
图1 亚甲二氧基取代的苯乙胺类β酮化合物的基本结构及几种典型的该类物质的分子结构
1 材料与方法
1.1 主要仪器与试剂
HP6890GC/5973MSD型气相色谱-质谱联用仪(美国Agilent公司),ACQUITY UPLC I-Class/Vion IMS QTof型超高效液相色谱/离子淌度-四极杆串联飞行时间质谱联用仪(美国Waters公司),JNM-ECS400型核磁共振仪(400MHz,日本电子株式会社),SB-5200DT型超声波清洗器[必能信超声(上海)有限公司],5415D型台式高速离心机(美国Eppendorf公司),Vortex-Genie2型涡旋振荡器(美国Scientific Industries公司)。
Dibutylone对照品(纯度>98%,法庭科学特种化学品联合实验室),甲醇(分析纯,上海虹口东风化学试剂有限公司),盐酸双苯戊二氨酯(SKF525A,美国Sigma-Aldrich公司),氘代甲醇(美国Sigma-Aldrich公司),乙腈、甲酸(质谱纯,美国Thermo Fisher Scientific公司),超纯水由Milli-Q Integral 3超纯水仪(美国Millipore公司)制备。
1.2 仪器条件
1.2.1 GC-MS
色谱条件:HP-5MS石英毛细管色谱柱(30 m×0.25mm,0.25μm,美国Agilent公司);柱温程序为起始100℃保持1min,以20℃/min升至280℃,保持15min;进样口温度260℃;载气为氦气(He),流速1.0mL/min,分流进样,分流比20∶1;进样量1μL。
质谱条件:电子轰击(electron impact,EI)离子源,电子能量70 eV;传输线温度280℃,离子源温度230℃,四极杆温度150℃;质量扫描范围m/z40~650;采用全扫描模式;溶剂延迟2.8 min。质谱结果与美国国家标准与技术研究院(National Institute of Standards and Technology,NIST)17.L谱库检索比对。
1.2.2 超高效液相色谱-四极杆飞行时间质谱(ultrahigh performance liquid chromatography-quadrupole time-of-flight mass spectrometry,UPLC-QTOF-MS)
色谱条件:ACQUITY UPLC BEH C18色谱柱(100 mm×2.1 mm,1.7 μm,美国Waters公司);以0.1%甲酸水溶液为流动相A,0.1%甲酸乙腈溶液为流动相B;采用梯度洗脱,洗脱程序见表1;柱温40℃;流速0.4mL/min;进样量1μL。
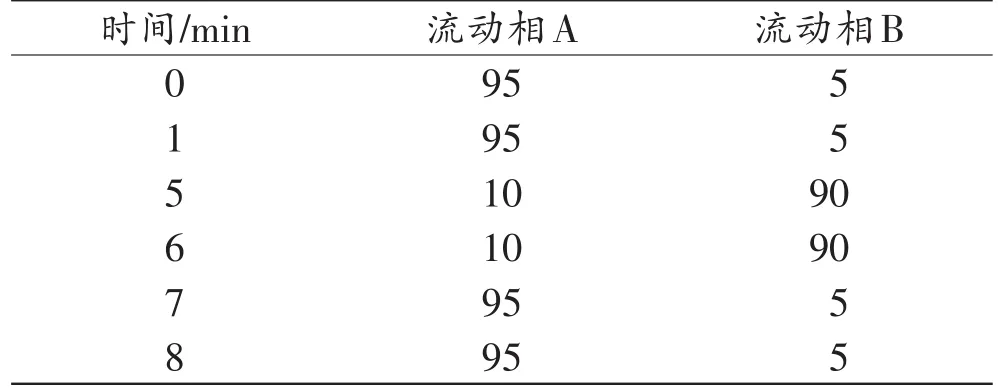
表1 梯度洗脱程序 (%)
质谱条件:电喷雾离子源(electrospray ioniza⁃tion,ESI)正离子模式,毛细管电压3.0 kV,离子源温度120℃,脱溶剂气温度550℃,锥孔反吹气流速50L/h,脱溶剂气流速800L/h,质量扫描范围m/z50~1 000,MSE扫描模式(低碰撞能量6 eV,高碰撞能量20~40 eV),以亮氨酸脑啡肽(m/z556.276 6)作为外标(LockSpray)进行质量数实时校正。
数据采集和分析:运用UNIFI 1.8.2.169软件采集数据和控制系统,结合Waters自带的筛查库和毒物库中的信息,并导入结构式mol文件,建立包含化合物名称、分子式、分子结构和精确质量数的目标数据库。将采集获得的原始数据导入UNIFI软件与目标数据库进行检索和比对,最终显示匹配度最高的结果。
1.2.3 核磁鉴定
以氘代甲醇溶液溶解样品,用1H NMR(400MHz)进行检测。
1.3 样品处理
实验样品为涉毒案件中缴获的粉色药片,每片质量约337mg。
取约10mg碾碎的药片置于10mL玻璃具塞试管中,加入适量甲醇溶液超声振荡10min充分溶解,以离心半径3.5 cm,12 000 r/min,离心1 min后,吸取上清液1mL,加入含内标SKF525A(0.02mg/mL)的甲醇溶液稀释至10 mL,取约1 mL装入自动进样小瓶内,供GC-MS;取约0.01mL装入自动进样小瓶内,用甲醇溶液稀释至1mL,供UPLC-QTOF-MS检测。
取3片药片碾碎用甲醇溶液充分浸泡溶解,过滤,用少量甲醇溶液润洗固体3遍。浓缩滤液,再经硅胶柱层析分离纯化,淋洗液为二氯甲烷/甲醇(梯度洗脱体积比为15∶1到10∶1),得到可疑组分的浅黄色产物约75mg。取约10mg样品,置于核磁管中,加入氘代甲醇溶液0.6 mL,使样品充分溶解,供1H NMR检测。
2 结 果
2.1 GC-MS检测
经GC-MS检测得到待测样品的总离子流色谱图和质谱图(图2~3)。由图可以看出,待测样品在保留时间为8.12 min和8.28 min处有两个主要组分,其中8.28min的组分经鉴定为咖啡因(一种中枢神经兴奋剂,也属于精神药品管控品种,常被用作毒品片剂中的添加剂)。另一未知组分的特征碎片离子(m/z)和相应的丰度比通过NIST 17.L谱库检索比对,得到与其特征碎片离子及丰度比都相似的匹配结果为bk-DMBDB,即Dibutylone。
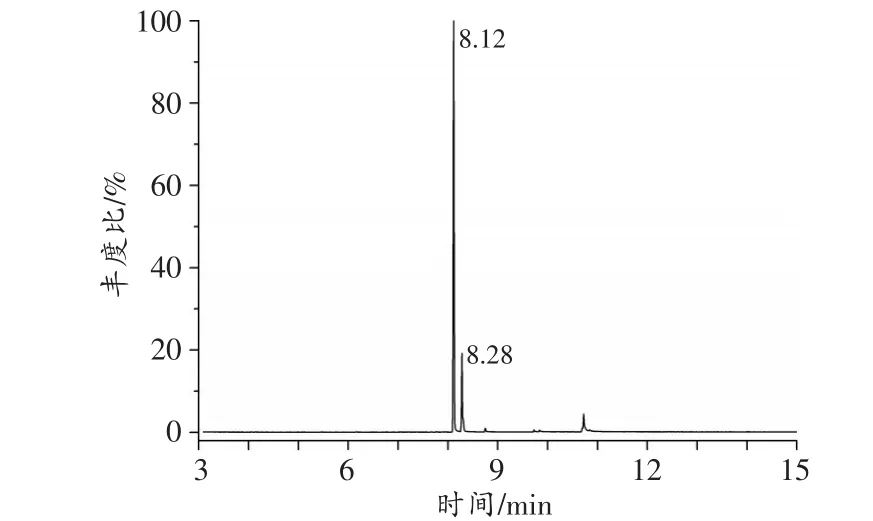
图2 待测样品的总离子流色谱图
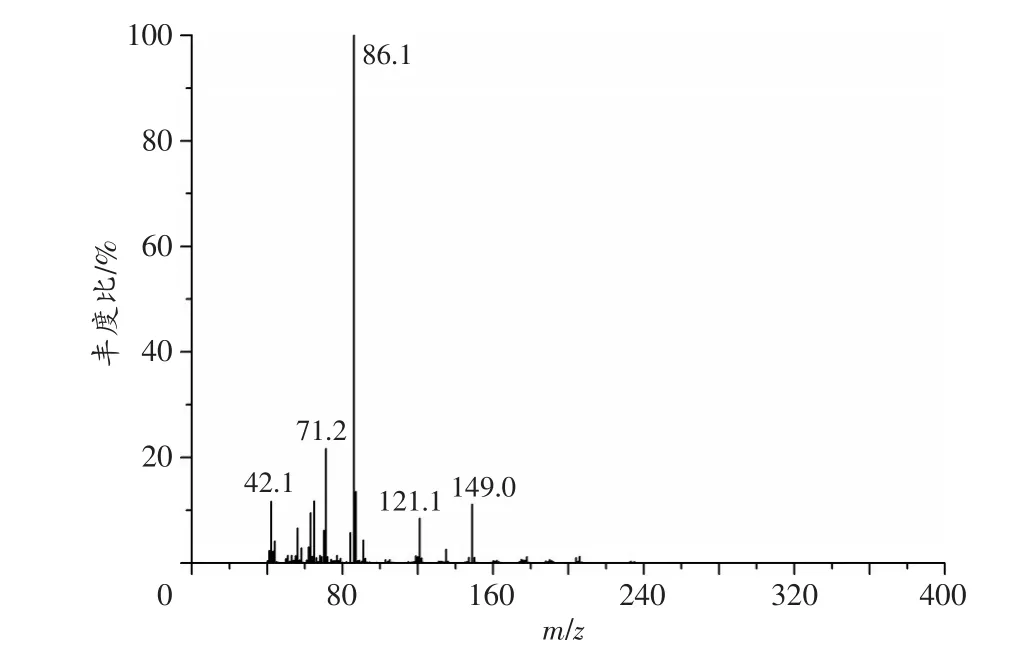
图3 待测样品中Dibutylone(经后续实验确证)的质谱图
2.2 UPLC-QTOF-MS检测
根据精确质荷比和碎片离子信息结合目标数据库对待测物进行结构鉴定,结果发现在保留时间2.61 min处的色谱峰与Dibutylone匹配一致,色谱图和质谱图见图4~5。该组分母离子和特征碎片离子的实测精确质荷比与Dibutylone的理论值偏差均不超过0.8×10-3,其主要碎片的离子信息见表2。
在高碰撞能量时的质谱图中可观察到明显的Di⁃butylone特征碎片m/z191.071 05、m/z161.060 45,因此推断该组分为Dibutylone。
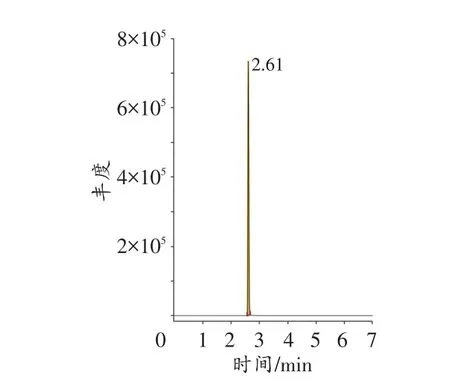
图4 待测物的提取离子流色谱图
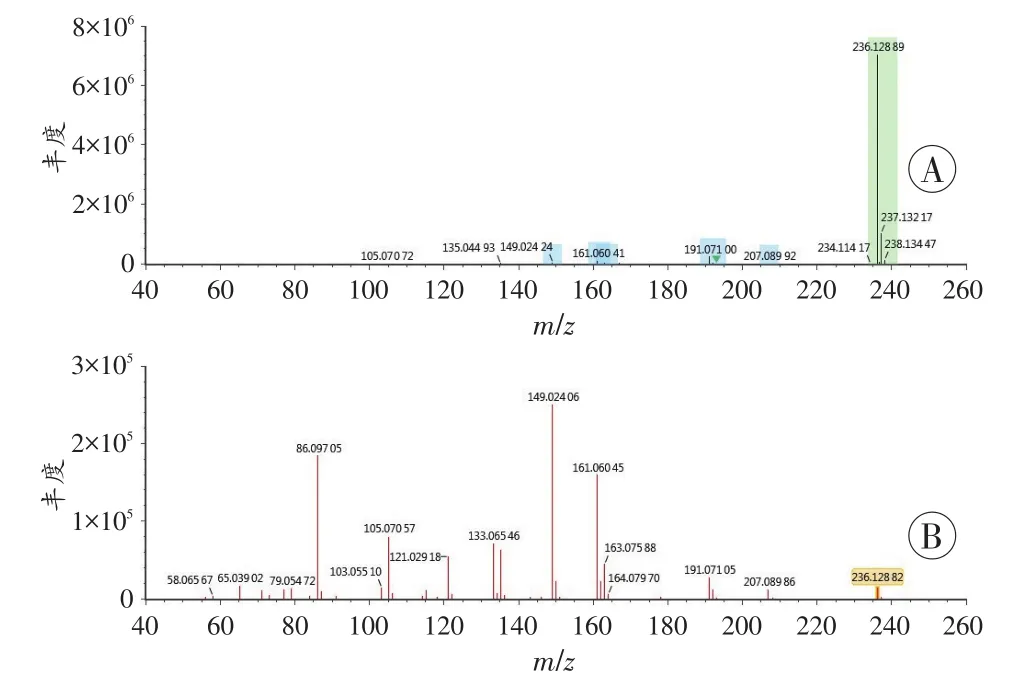
图5 待测物的高分辨质谱图
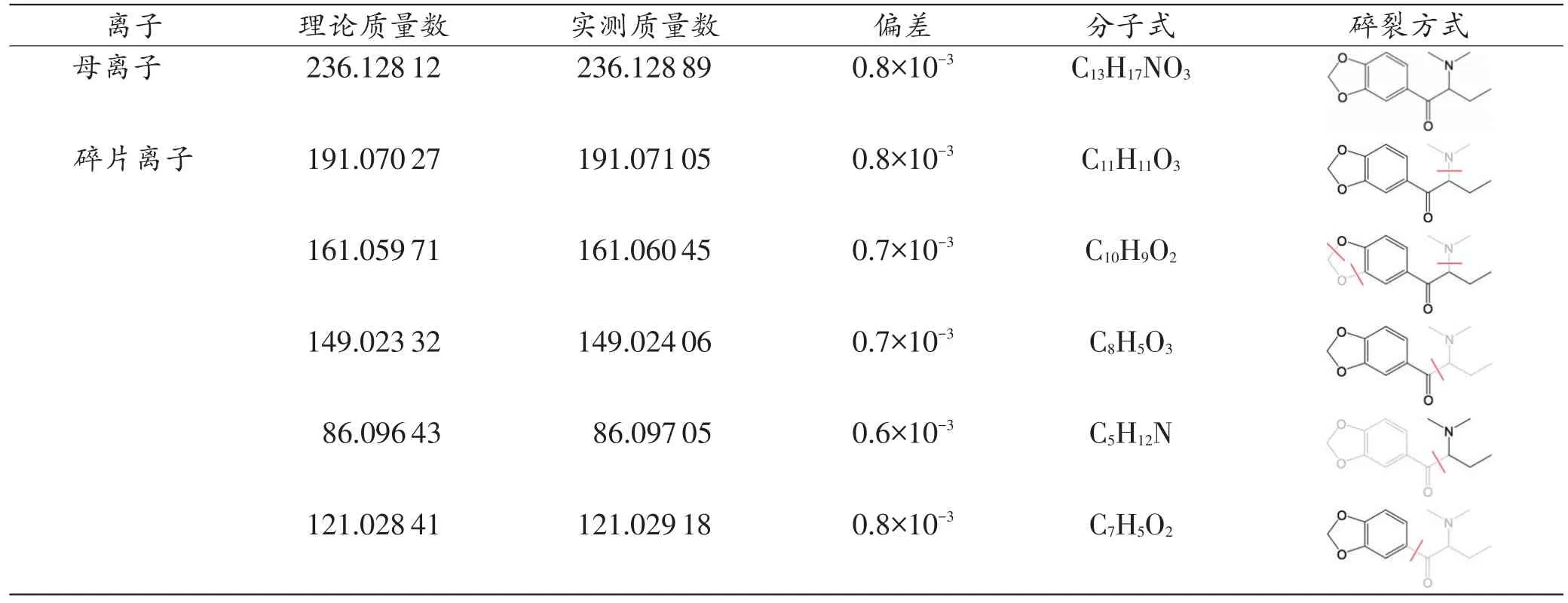
表2 待测物目标组分的离子信息
2.3 核磁鉴定结果
为了进一步验证待测样品中可疑组分的结构,进行了1H NMR检测。由于待测样品为混合物,所以先采用硅胶柱层析进行分离纯化,纯化得到浅黄色固体粗产物约75mg。核磁鉴定结果见图6、表3,其中δ3.31、δ4.89为CD3OD的溶剂峰和水峰。1H NMR:δ0.85(3H,CH2CH3),δ2.05~2.21(2H,NCHCH2CH3),δ2.88(3H,NCH3),δ3.00(3H,NCH3),δ5.20(1H,NCHCH2),δ6.14(2H,OCH2O),δ7.03、δ7.53、δ7.76(3H,C6H3)。核磁的鉴定结果验证了该可疑组分为Dibutylone。
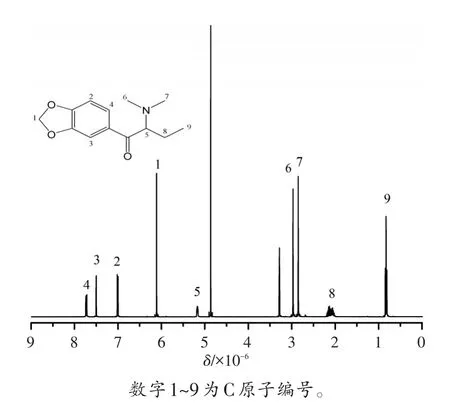
图6 可疑组分的1H NMR图谱
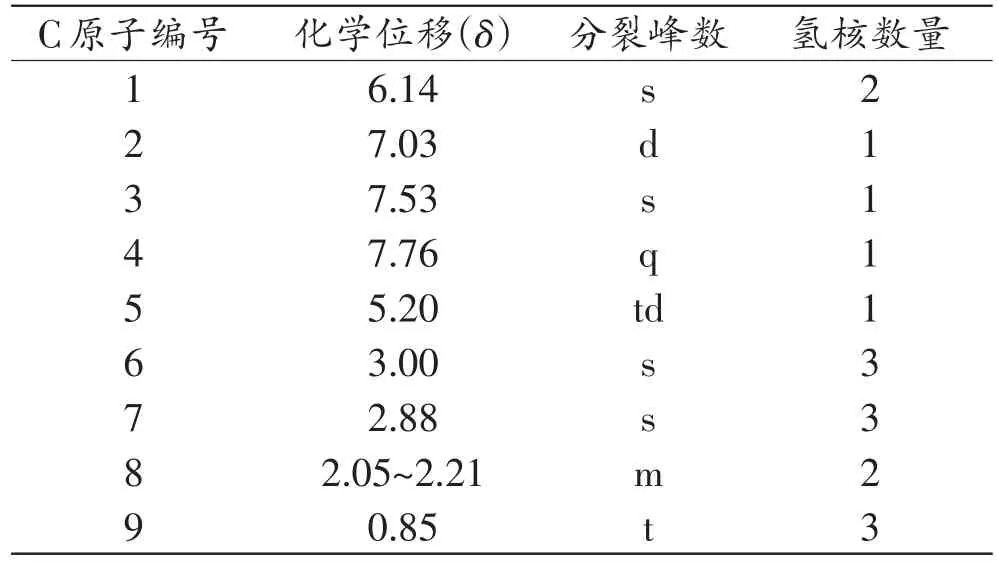
表3 可疑组分1H NMR的数据信息
2.4 对照品比对结果
本鉴定中心委托上海市法庭科学特种化学品联合实验室合成了Dibutylone对照品。用甲醇溶液将对照品配制成质量浓度为0.50mg/mL的溶液,按1.2.1节条件进行GC-MS检测。结果表明,待测样品中未知组分的保留时间和特征碎片离子均与对照品一致,可确证为Dibutylone。
3 结 论
本研究建立了GC-MS、高分辨质谱结合谱库检索技术筛查新精神活性物质Dibutylone的方法。高分辨质谱能得到目标物的精确质量数,是进行未知物鉴定的常用手段。本研究的高分辨质谱条件运用了MSE的扫描方式,由“低碰撞能量”与“高碰撞能量”两种扫描交替构成,能在一次分析中同时获得母离子与碎片离子的精确质量数,再结合特有数据库的筛查功能,可大大提高定性分析的准确度。
本研究还运用核磁共振波谱技术对待测物的结构进行了进一步确证。尽管核磁共振波谱技术是对化合物进行结构鉴定的有力手段,但一般适用于检测纯度较高的样品。本研究中的待测样品为含多组分的混合物,首先采用硅胶柱层析技术对各组分进行分离纯化,收集并浓缩以获得较高纯度的可疑组分,再用于后续的核磁分析。
猜你喜欢
现代仪器与医疗(2022年4期)2022-10-08
农业工程学报(2022年6期)2022-06-27
山东化工(2020年15期)2020-09-01
实验室研究与探索(2020年6期)2020-08-25
铜仁学院学报(2018年6期)2018-07-05
安阳工学院学报(2016年6期)2016-12-06
化工生产与技术(2016年5期)2016-11-07
百姓生活(2016年6期)2016-06-22
中国洗涤用品工业(2016年2期)2016-02-28
中国塑料(2015年2期)2015-10-14
