
Cardiac amyloidosis:A case report and review of literature
2019-04-17 02:25AdeyemiAdedamolaTaiwoLavanyaAlapatiAssadMovahed
World Journal of Clinical Cases 2019年6期
Adeyemi Adedamola Taiwo,Lavanya Alapati,Assad Movahed
Abstract
Key words: Cardiac amyloidosis;Restrictive cardiomyopathy;Transthyretin;Case report
INTRODUCTION
With an increasingly ageing population and advances in cardiovascular imaging techniques,conditions that previously went undiagnosed and thought to be uncommon are being increasingly identified.Cardiac amyloidosis is one of such conditions.In addition,with broad research ongoing on various modalities of treatment,early diagnosis may be crucial to limiting further amyloid deposition and thus,alter the disease course.In this case report,we discuss a 73 years old female admitted in cardiogenic shock with subsequent workup confirming a diagnosis of cardiac amyloidosis.A review of the condition focusing on the diagnosis and treatment is also included.
CASE PRESENTATION
Chief complaints
A 73 years old African American female presented to the emergency department with sudden onset shortness of breath with no associated chest pain.She was emergently intubated due to impending respiratory failure.
History of present illness
Her medical history was notable for congestive heart failure,atrial fibrillation,primary hyperparathyroidism,carpal tunnel syndrome and spinal stenosis.
History of past illness
She had a longstanding history of heart failure first diagnosed in January 2010 with echocardiogram (ECHO) demonstrating a left ventricular ejection fraction (LVEF) of 40%.A cardiac catheterization in February 2014 revealed non-obstructive coronary artery disease;hence,the cardiomyopathy was labelled non-ischemic.Due to nonrecovery of systolic function and persistent left bundle branch block (LBBB),she had a biventricular cardioverter-defibrillator (ICD) implanted in March 2014.
Medication History
Her pre admission medications included apixaban 5 mg twice a day,sacubitrilvalsartan (24-26 mg) twice a day,furosemide 20 mg twice a day,metoprolol succinate 25 mg daily and sotalol 80 mg twice a day.
Physical examination upon admission
On initial physical examination,she had a blood pressure of 80/54 mmHg with pulse rate of 75 beats per minute (bpm).Her jugular venous pressure was not elevated.She had clear lungs and normal heart sounds with no murmurs or gallops on auscultation.Her lower extremities were cool to touch with mild pitting edema bilaterally.
Laboratory examinations
Laboratory testing was significant for N-terminal pro b-type natriuretic peptide (NTProBNP) elevation at 12914 pg/mL (Normal < 125 pg/mL).Complete blood count and comprehensive metabolic panel were within normal limits.
Serum protein electrophoresis obtained to evaluate for an underlying gammopathy(seen in light chain amyloidosis) demonstrated moderate hypoproteinemia and hypoalbuminemia with no monoclonal spike (M spike).The serum free kappa to lambda light chain ratio was within normal limits and urine immunofixation was negative for immunoglobulin light chain ruling out a gammopathy.
Imaging examinations
Electrocardiogram (ECG) showed LBBB and low voltages in the limb leads (Figure 1)while the chest radiograph demonstrated bilateral patchy opacities consistent with pulmonary edema.
Transthoracic ECHO revealed a borderline dilated left ventricle (LV) with severe left ventricular hypertrophy and reduced LVEF of 30%-35% (Figures 2-6).
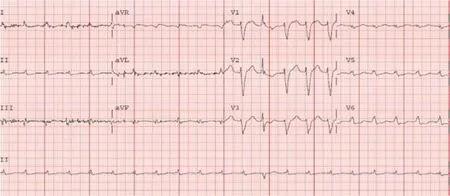
Figure1 Electrocardiogram - Left bundle branch block with low voltage in limb leads.
Due to the discordance between the degree of LV thickness on ECHO and QRS voltage on ECG,cardiac amyloidosis was considered as a possible etiology of the cardiomyopathy.
Cardiovascular magnetic resonance imaging (MRI) could not be obtained as her ICD was not MRI compatible.A99Technetium pyrophosphate (99mTc-PYP) planar scintigraphy was subsequently done (Figure 7).Quantitative analysis of heart retention patterns showed a heart to contralateral (H/Cl) ratio of 1.8,strongly suggestive of transthyretin amyloid deposition.An abdominal fat pad biopsy was also done which had no evidence of amyloid deposition on pathologic examination.
Medical management
A pulmonary artery catheter was placed on admission which showed a central venous pressure of 3 mmHg with cardiac output (CO) of 2.06 L/min and cardiac index of 1.12 L/min/m2.An assessment of cardiogenic shock was made and inotrope therapy with intravenous dobutamine was initiated.
With improvement in her hemodynamic status,she was able to be weaned off the inotrope support and was eventually extubated.Her hospital course was complicated by multiple episodes of flash pulmonary edema.This was attributed to the narrow window between high and low filling pressures which made accurate assessment of fluid status and diuretic requirements tedious.She progressed well with addition of isosorbide dinitrate/hydralazine (40-75 mg) twice a day to her medication regimen prior to discharge home.
Post discharge,an endomyocardial biopsy was done which confirmed Transthyretin Amyloidosis - wild type.
FINAL DIAGNOSIS
Wild type transthyretin cardiac amyloidosis.
TREATMENT
The main aim of treatment following diagnosis was optimizing her cardiac function and managing symptoms as there is no approved therapy for transthyretin cardiac amyloidosis currently.Neurohormonal blocking agents for her systolic dysfunction were continued with titration of her diuretic depending on her volume status.In the future,she may be a good candidate for one of the amyloid specific therapies under investigation.
OUTCOME AND FOLLOW-UP
She has been enrolled in an amyloid program and has required regular clinic follow up since diagnosis.She has remained stable.
DISCUSSION
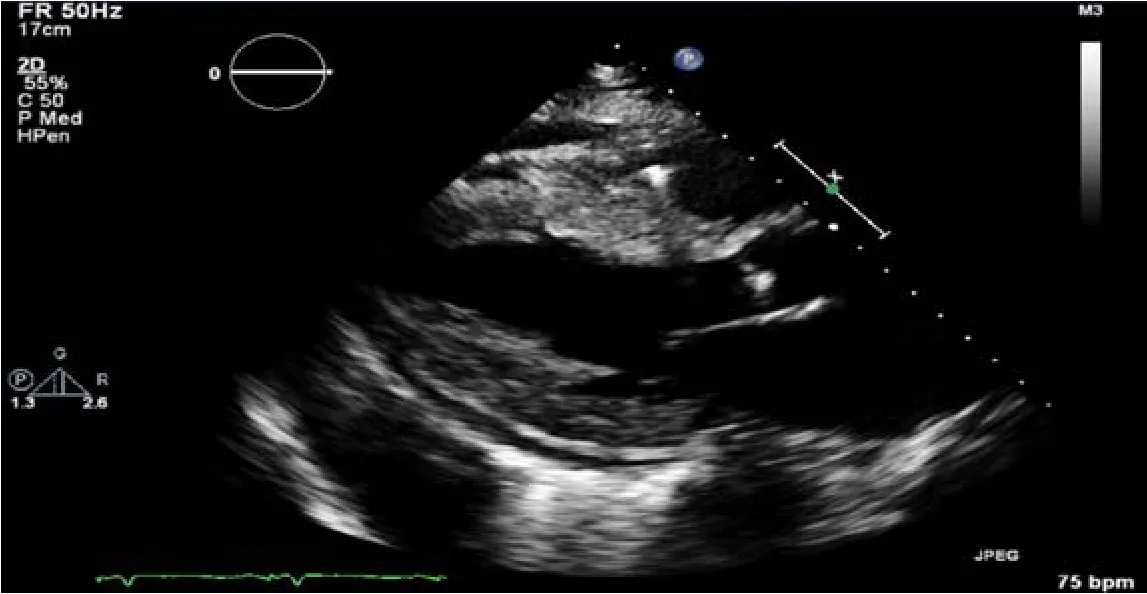
Figure2 Parasternal long axis echocardiographic view demonstrating severe concentric left ventricular hypertrophy with small pericardial effusion.
Amyloidosis is a systemic disorder characterized pathologically by the extracellular deposition of insoluble fibrils composed of misfolded proteins in various organs including the heart with resulting alteration in structure and function[1].The amyloid fibrils are rigid,proteolytic resistant structures,typically less than 10 nanometers in diameter with a characteristic apple-green birefringence with Congo-red staining under polarized light microscopy[2].
Amyloidosis may be hereditary or acquired and the nature of the precursor protein of the fibrils forms the basis of classification[3].More than 30 such precursor proteins have been identified and of these,2 types are responsible for about 95% of cases of cardiac amyloidosis:Immunoglobulin light chain amyloidosis (AL) and transthyretin amyloidosis (ATTR)[4].
AL,although overall a rare condition,is the most frequently diagnosed form with about 0.3 cases per 100000 population in a large referral center[5].AL occurs secondary to a plasma cell dyscrasia resulting in excessive production of immunoglobulin light chain units,with associated misfolding and deposition in extracellular tissue.In addition to cardiac manifestations,hepatic,renal and neurologic complications may be seen in AL.Early diagnosis and initiation of treatment in AL may alter disease outcome although mortality remains high[6].
ATTR occurs due to the misfolding of the liver derived precursor protein transthyretin (TTR,previously called prealbumin).TTR has a tendency to dissociate into dimers and monomers and subsequently aggregate into insoluble fibrils[7,8].Two variants of this form of amyloidosis have been described - the hereditary/mutant form caused by mutations in theTTRgene and the sporadic/wild type (systemic senile amyloidosis)[9].
The phenotype of mutant ATTR is dependent on the nature of the underlying mutation with over 100 different mutations known to involve the TTR gene.The Val30Met (Valine to Methionine at position 30) mutation is the most common worldwide[7].Of particular significance from a cardiac standpoint is the Val122Ile(Valine to Isoleucine at position 122) mutation which has been identified in a significant percentage of the North American black population and is associated with an increased risk of cardiac manifestations.Neurologic symptoms in the form of peripheral/autonomic neuropathy are the dominant presentation of mutant ATTR.
Wild type ATTR was previously described to be exclusive to older white males.However,more cases in females are being identified as in our patient suggesting this condition may underdiagnosed in this population[2,7].Initial manifestations of wild type ATTR may include carpal tunnel syndrome (especially bilateral) and spinal stenosis and these may precede the cardiac complications by several years.
Pathology/pathophysiology
The diffuse deposition of amyloid fibrils in the cardiac interstitial space produces significant thickening of both ventricles with associated stiffness which results in impaired diastolic relaxation and the characteristic restrictive physiology[2,9].Fibril deposition within and/or around the smaller vessels of the heart with resulting narrowing may produce ischemia/infarction[10].In addition,there is evidence pointing to the direct toxicity of the fibrils to the cardiac myocytes especially with AL[9,11].The atria,valves and conduction system are also typically involved resulting in sequelae such as atrial fibrillation,valvular regurgitation and various degrees of heart block.
Clinical manifestations

Figure3 Short axis echocardiographic view demonstrating severe concentric left ventricular hypertrophy with small pericardial effusion.
The classic clinical presentation of cardiac amyloidosis is with symptoms of congestive heart failure including exertional dyspnea and orthopnea.Fatigue and weakness are also common,resulting from compromised CO.Symptoms of right ventricular failure including abdominal distension and lower extremity edema may be more prominent in certain patients[2,12].
Coronary vessel involvement may manifest with angina while palpitations and syncope may be seen with the various conduction abnormalities associated with this condition,atrial fibrillation being the most commonly identified arrhythmia[2,12].
Pericardial and pleural effusions may be seen with more advanced cardiac dysfunction.A great variety of extra-cardiac symptoms/signs may be seen in amyloidosis including ecchymoses/petechiae,hepatomegaly,macroglossia,nonspecific gastrointestinal symptoms such as diarrhea/weight loss and carpal tunnel syndrome (especially bilateral which is common in ATTR)[13,14].
Clinical evaluation
Diagnosing cardiac amyloidosis requires a high index of suspicion due to its relatively low incidence.Typically,a combination of laboratory tests,ECG,cardiac imaging and histopathology is required.
Plasma levels of the natriuretic peptides (BNP and NT-proBNP) tend to be high in cardiac amyloidosis irrespective of severity of heart failure.This likely reflects the direct toxicity of the amyloid fibrils at a cellular level as alluded to previously[15].Measuring the level of NT-proBNP however has some prognostic value with its use in the Mayo staging system for AL.
Serum levels of troponin T and/or troponin I are frequently chronically elevated in CA.This may reflect small vessel ischemia or direct myocyte toxicity[2].Takashioet al[16]however demonstrated the utility of high sensitivity cardiac troponin T in differentiating cardiac amyloidosis from other cardiac conditions with a hypertrophic phenotype.
Assaying circulating levels of TTR has limited clinical utility in evaluating for ATTR.Work up for a monoclonal process to evaluate for AL is however always indicated when cardiac amyloidosis is suspected.The preferred test for this is the serum free light chain assay with immunofixation (as against the traditional serum and urine protein electrophoresis which are not sufficiently sensitive)[2].An abnormal kappa to lambda ratio (< 0.26 or > 1.65) in combination with a M protein spike on immunofixation is highly suggestive of a monoclonal light chain process[2].It is important to note that identifying an abnormal plasma cell clone alone is insufficient for diagnosis as a moderate increase in circulating light chains in not necessarily pathologic.
The first clue which may prompt suspicion for cardiac amyloidosis is the ECG.The archetypal finding is low QRS voltages due to the electrically silent amyloid fibrils which are not detected by the ECG[2,12].This is inconsistent with the degree of ventricular thickening seen on ECHO.The absence of low QRS voltage does not preclude a diagnosis of cardiac amyloidosis with only 30%-70% of patients meeting low voltage criteria on ECG[17,18].A pseudoinfarct pattern with Q waves in the early precordial leads is another common ECG finding,identified in about 40%-50% of CA patients[19,20].Other ECG findings frequently encountered include atrial arrhythmias such as atrial fibrillation,various degrees of atrioventricular block,interventricular conduction delays and bundle branch blocks.

Figure4 Four chamber echocardiographic view demonstrating bi atrial enlargement,left ventricular hypertrophy,small pericardial effusion and implantable cardioverter-defibrillator device.
The ECHO findings in cardiac amyloidosis are nonspecific but highly suggestive in the right clinical context.The hallmark finding is symmetric biventricular wall thickening with a spectrum of diastolic abnormalities varying from abnormal relaxation to a restrictive filling pattern[2,9,21].Cardiac amyloidosis should be suspected with LV wall thickness greater than 12 mm in the absence of preceding history of hypertension[2].Atrial dilation is commonly seen due to the combination of deposition of fibrils in the atria and elevated LV filling pressures.Thickening of the heart valves is also commonplace which helps to distinguish cardiac amyloidosis from hypertensive heart disease[2].LVEF typically remains preserved until later in disease course when the effects of myocyte necrosis and fibrosis affect systolic function[21].An apical sparing pattern of strain on 2D speckle ECHO has been described by several authors as being specific for cardiac amyloidosis enabling differentiation from other cardiac diseases with a hypertrophic phenotype[2,21].The pattern of strain is due to a decrease in longitudinal strain in the basal and mid wall regions in comparison to the apex where the strain is relatively preserved[12].Small pericardial effusions are common and nonspecific.Larger effusions and cardiac tamponade are relatively rare.
Cardiac MRI has taken on an increasingly important role in the screening and evaluation of cardiac amyloidosis.The typical finding is a transmural or subendocardial pattern of late gadolinium enhancement (LGE).Global involvement is more commonly encountered although a focal patchy pattern may be seen.These features may precede the morphologic phenotype of increased LV wall thickness[22-24].In AL cardiac amyloidosis,the global pattern of LGE has been shown to be predictive of mortality and provides prognostic information beyond the clinical and biochemical data used in the Mayo staging[25].The main constraint to use of this imaging modality is the inability to administer gadolinium contrast to patients with significant kidney disease.
Nuclear scintigraphy with the use of bone seeking,phosphate-based radiotracers including99mTc-PYP and99mTc-DPD (technetium-3,3-diphosphono-1,2-propanodicarboxylic acid) has demonstrated excellent sensitivity and specificity in diagnosing cardiac amyloidosis and differentiating ATTR from AL[26,27].Only99mTc-PYP is currently approved for use in the United States but similar results have been demonstrated with99mTc-DPD in European studies.The exact mechanism for accumulation of the radiotracers in ATTR cardiac amyloidosis is unclear.It is hypothesized that this may be related to higher calcium levels in ATTR which results in more intense binding with the radiotracers[26].By comparing the heart retention patterns with the contralateral chest wall,a heart to contralateral ratio is obtained.A heart to contralateral ratio greater than 1.5 is strongly suggestive of ATTR cardiac amyloidosis.
There has been recent increased interest in the role of positron emission tomography/computerized tomography (PET/CT) in the imaging of cardiac amyloidosis.PET tracers,fluoride labelled radioisotopes including18F-florbetapir and18F-florbetapen,have been demonstrated to bind at sites of amyloid deposition irrespective of the nature of the precursor protein[28].The potential utility of this imaging modality has been evaluated in a few small single center studies.Using parameters including the standard uptake value and retention index,subjects with both ATTR and AL cardiac amyloidosis were accurately identified over controls and the burden of amyloid deposition could be quantified[29,30].Larger,multicenter studies are required to evaluate the clinical utility of this modality.

Figure5 Doppler demonstrating restrictive physiology with medial E’ velocity of 2.78 cm/s and E/Med E’ 39.2.
Despite these advances in cardiovascular imaging,histopathologic confirmation of amyloid deposits in tissue remains the gold standard.In addition to confirming the diagnosis,it also allows identification of the specific precursor protein which is important for guiding therapy[4].Tissue may be obtained from extra-cardiac sites such as the abdominal fat pad and rectum by fine needle aspiration and apple green birefringence under polarized light microscopy after Congo-red staining is diagnostic.Although immunohistochemical techniques may be used,liquid chromatographytandem mass spectrometry on samples obtained by laser microdissection is a more sensitive and specific test for identifying the specific amyloid protein[4].A negative biopsy from an extra cardiac site does not rule out cardiac amyloidosis and an endomyocardial biopsy should be obtained in such cases.This procedure is however limited to larger referral centers and carries the small but significant risk of ventricular perforation.
Treatment
The treatment of cardiac amyloidosis is two pronged - treatment of symptoms arising from cardiac dysfunction and treatment of the underlying amyloidosis.The most commonly encountered manifestations of cardiac amyloidosis are congestive heart failure (CHF) and conduction system abnormalities.
CHF in cardiac amyloidosis is most commonly diastolic in nature (heart failure with preserved ejection fraction) with systolic dysfunction occurring late in disease course.As a result,there is a distinct lack of therapies with demonstrated mortality benefit.The cornerstone of treatment remains restricting salt and fluids in addition to the judicious use of diuretics[2,12].A combination of loop diuretics and aldosterone receptor antagonist appears to work best although close monitoring of volume status is required as CA patients are preload dependent[12,31].Traditional therapies for systolic heart failure such as beta-blockers and angiotensin converting enzyme inhibitors are avoided due to risks of hypotension and renal injury.
Atrial fibrillation is the most common arrhythmia encountered[19]and usual rate controlling medications including beta blockers and calcium channel blockers are poorly tolerated due to their hypotensive side effects.Digoxin is also best avoided due to increased risk of toxicity from binding to the amyloid fibrils[32].Amiodarone is a commonly used option and is relatively well tolerated[9].Cardiac amyloidosis patients are at increased risk of intracardiac thrombus formation (especially AL) and this risk is further exacerbated in setting of atrial fibrillation.Anticoagulation is thus indicated with some centers recommending anticoagulation even in normal sinus rhythm if atrial mechanical dysfunction is present[33].Permanent pacemaker implantation is indicated for symptomatic therapy in patients with higher degree heart blocks.There is limited evidence for the mortality benefit of ICD in primary prevention as sudden death most commonly occurs from bradycardia and pulseless electrical activity[34,35].
There are 2 main approaches to treating the underlying amyloidosis - stopping further production of the precursor protein and reducing the burden of already deposited amyloid.
In AL,therapy is directed towards the plasma cell clone with chemotherapy followed by autologous hematopoietic stem cell transplant (HSCT)[21].The most commonly used chemotherapy regimen involves the use of bortezomib,dexamethasone and an alkylating agent - typically cyclophosphamide which has demonstrated benefit in lower risk patients[6].A significant proportion of patients with AL have advanced cardiac disease at the time of diagnosis which limits their ability to receive high dose chemotherapy and HSCT.
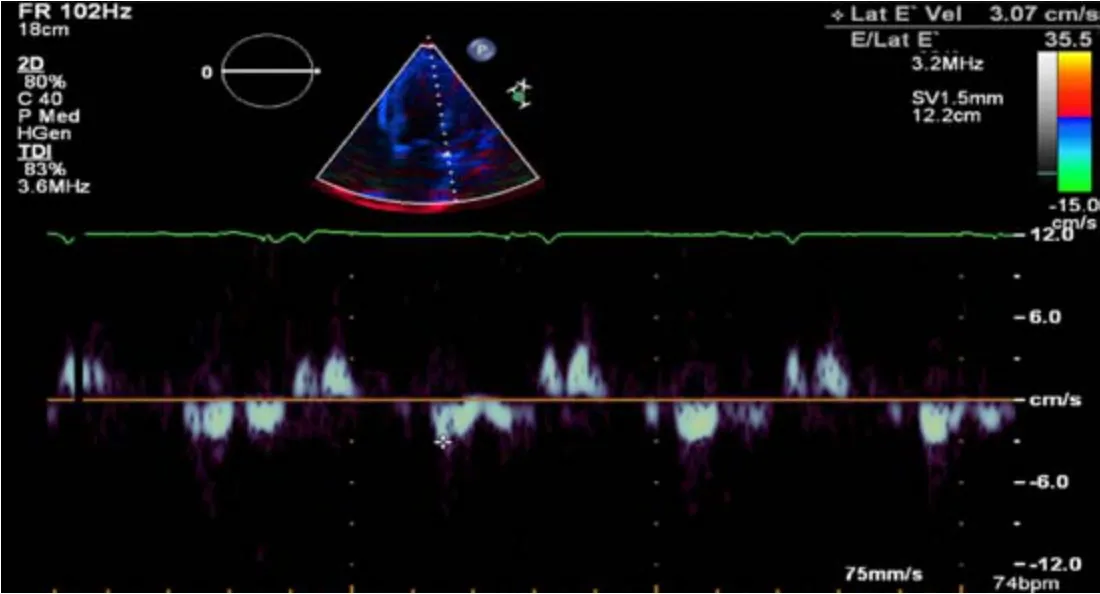
Figure6 Doppler demonstrating restrictive physiology with lateral E’ velocity of 3.07cm/s and E/Lat E’ 35.5.
With the liver being the primary site of TTR production,orthotropic liver transplantation (OLT) has been used as a curative strategy for the treatment of mutant ATTR.Although this brought about an improvement in neurologic symptoms,Dubreyet al[36]noted a progression in ventricular thickening despite elimination of TTR production.This has been hypothesized to be due to the deposition of wild type TTR on the pre-existing focus of mutant TTR[37].OLT is not validated in the treatment of wild type ATTR.
Several interventions aimed at decreasing the production of TTR are at various stages of development.In August 2018,the Food and Drug Administration (FDA)approved patisiran for the treatment of polyneuropathy associated with mutant ATTR after meeting the primary end point in the APOLLO clinical trial[38].Patisiran is a small interfering ribonucleic acid that targets the TTR messenger ribonucleic acid(mRNA) and decreases gene expression[2,9].On analysis of the APOLLO study by Solomonet al[39],patisiran was also noted to decrease cardiac parameters such as the mean LV wall thickness and global longitudinal strain with improvement in CO indicating a potential role in the treatment of ATTR cardiac amyloidosis.
In a similar vein,the anti-sense oligonucleotide inotersen was approved by the FDA for the treatment of polyneuropathy associated with mutant ATTR in October 2018.It decreases the production of TTR by binding the mRNA and causing its degradation[2].Further studies on its effect on cardiac disease are needed.
An alternative approach to treating ATTR involves the use of TTR tetramer stabilizers (such as tafamidis) which bind at specific sites on the TTR molecule and stabilize its tetrameric structure preventing further amyloid fibril formation[2].A recently completed phase 3 trial (ATTR-ACT trial) demonstrated the benefits of tafamidis in ATTR cardiac amyloidosis with reductions in all-cause mortality and cardiovascular related hospitalizations in both the mutant and wild-type disease[40].Tafamidis has been designated a breakthrough therapy by the FDA and is currently awaiting approval.
Diflunisal,a non-steroidal anti-inflammatory drug stabilizes the TTR molecule in a similar manner preventing amyloid fibrillogenesis.It demonstrated its utility in decreasing the rate of neurologic deterioration in patients with neuropathic manifestations of mutant ATTR[41].Its use in advanced cardiac disease is however limited by side effects such as fluid retention and renal insufficiency.
A third approach towards treatment of ATTR cardiac amyloidosis involves the use of agents that disrupt and facilitate clearance of already deposited fibrils.The tetracycline antibiotic doxycycline in combination with tauroursodeoxycholic acid,a biliary acid,has demonstrated some utility in this role in mouse models but human studies are limited[42].
The role of orthotropic heart transplantation (OHT) in the management of cardiac amyloidosis remains controversial due to concerns about recurrence of disease in transplanted hearts and long-term patient survival in an era with limited donors[43].It however may be the only treatment option for many patients.In AL cardiac amyloidosis,patients are only considered for transplant if they have limited extracardiac involvement and have demonstrated good response to initial chemotherapy (with expectation that the plasma cell clone can be controlled)[2,12].In ATTR cardiac amyloidosis,combined OHT and OLT may be considered in patients without other comorbidities[9,43].
Prognosis

Figure7 Planar scintigraphy using 99Technetium pyrophosphate for detection of amyloid protein in the heart.
Despite advancements in the diagnosis and treatment of cardiac amyloidosis,the overall prognosis remains poor.In untreated patients,ATTR cardiac amyloidosis has a slowly progressive course and a better prognosis than AL cardiac amyloidosis.The nature of the precursor protein (wild type vs mutant transthyretin) influences disease progression and outcomes[44].In wild type ATTR cardiac amyloidosis,the median survival from the time of diagnosis is reported to be about 4 years with the primary cause of death most commonly cardiovascular in nature[45,46].Predictors of reduced survival include advanced age,elevated BNP,decreased LVEF and increased relative wall thickness[45,46].In mutant ATTR cardiac amyloidosis,outcomes are dependent on the nature of the underlying mutation.The Val122Ile mutation is associated with worse outcomes with a median survival from diagnosis of about 2 years[47].
In untreated AL cardiac amyloidosis,median survival is about 6 months from the onset of heart failure[48].In patients with early stage disease who undergo HSCT,reduced post-transplant mortality and improved survival has been demonstrated with 4 year overall survival of about 90%[49,50].
CONCLUSION
Cardiac amyloidosis remains a rare disease despite a recent uptick in diagnoses owing to increased physician suspicion and improved cardiovascular imaging techniques.With emerging therapies that have the potential to improve patient outcomes,early diagnosis has taken on an increasingly important role.This case report discusses the clinical evaluation of cardiac amyloidosis highlighting the utility of various laboratory tests and imaging modalities in arriving at a diagnosis.
 World Journal of Clinical Cases2019年6期
World Journal of Clinical Cases2019年6期
- World Journal of Clinical Cases的其它文章
- Effects of apoptosis on liver aging
- Liver involvement in the drug reaction,eosinophilia,and systemic symptoms syndrome
- Surgical method choice and coincidence rate of pathological diagnoses in transduodenal ampullectomy:A retrospective case series study and review of the literature
- lndividualized minimally invasive treatment for adult testicular hydrocele:A pilot study
- Successful totally transthoracic echocardiography guided transcatheter device closure of atrial septal defect in pregnant women
- Successful treatment with hysteroscopy for infertility due to isthmocele and hydrometra secondary to cesarean section:A case report