
利用CRISPR/Cas9系统建立内生链霉菌SAT1的基因簇敲除体系
2019-07-26 09:10田文佳窦桂铭王莎孙靖雅马玉超
生物技术通报 2019年6期
田文佳 窦桂铭 王莎 孙靖雅 马玉超
(1. 北京林业大学生物科学与技术学院,北京 100083;2. 北京林业大学林学院,北京 100083)
内生链霉菌能够产生次级代谢产物抑制植物病原菌的生长,也能通过溶磷、固氮等途径促进植物生长,这些特点使其在现代农业的生物防治领域展现出广阔的应用前景[1]。同时,链霉菌的基因组(5.96-11.94 Mb)较一般细菌的基因组大,其基因组通常含有十几到几十个次级代谢基因簇,且单个基因簇长达十几甚至上百kb。庞大的基因簇及复杂的调控过程为链霉菌抑菌机制的研究和新的活性物质的发现增加了难度,也减缓了其在农林业领域应用的速度[2]。
全基因组测序技术和素有“基因魔剪”之称的CRISPR/Cas9为链霉菌天然产物的深入研究提供了强大的助力[3]。通过全基因组测序和生物信息学分析,我们可以了解链霉菌次级代谢基因簇的数量和类型,预测次级代谢产物的结构;通过CRISPR/Cas9技术对基因簇进行编辑,可以深入研究基因簇的功能和活性物质的生物合成过程。与传统的基因编辑方法相比,CRISPR/Cas9具有实验周期短、编辑片段长度灵活(既可以是长片段,也可以是单个核苷酸)、可以对基因组进行多次编辑等优点[4]。因此,该系统现已发展成为优良的基因编辑工具,并迅速成为生物科学和医学领域的研究热点[5]。近年来,该系统已成功应用于Bacillus、Clostridium、Corynebacterium、Escherichia coli、Lactobacillus、Mycobacterium、Pseudomonas、Staphylococcus 和Streptomyces等体系[6-14]。为了利用 CRISPR/Cas9技术对链霉菌基因组进行编辑,Cobb等[15]于2014年构建了基因敲除载体pCRISPomyces-2(图1)。该载体包含以下几部分元件:sgRNA和密码子优化的cas9基因,lacZ基因侧翼的Bbs I酶切位点,单一的Xba I酶切位点,安普霉素抗性基因[aac(3)-IV],大肠杆菌复制起点colE1,RP4接合转移原件oriT和pSG5的温度敏感rep区。利用pCRISPomyces-2对白色链霉菌的sshg_05713(67 bp)和sshg_00040/sshg_00050基因(13214 bp)以及绿色产色链霉菌的phpD(23 bp)和phpM基因(20 bp)进行敲除,效率高达66%-100%[15]。该系统的构建为链霉菌多个复杂基因簇的功能研究和新结构化合物的发现提供了新技术和新方法,质粒的快速构建也为敲除基因组上任何目标位点提供了方便。
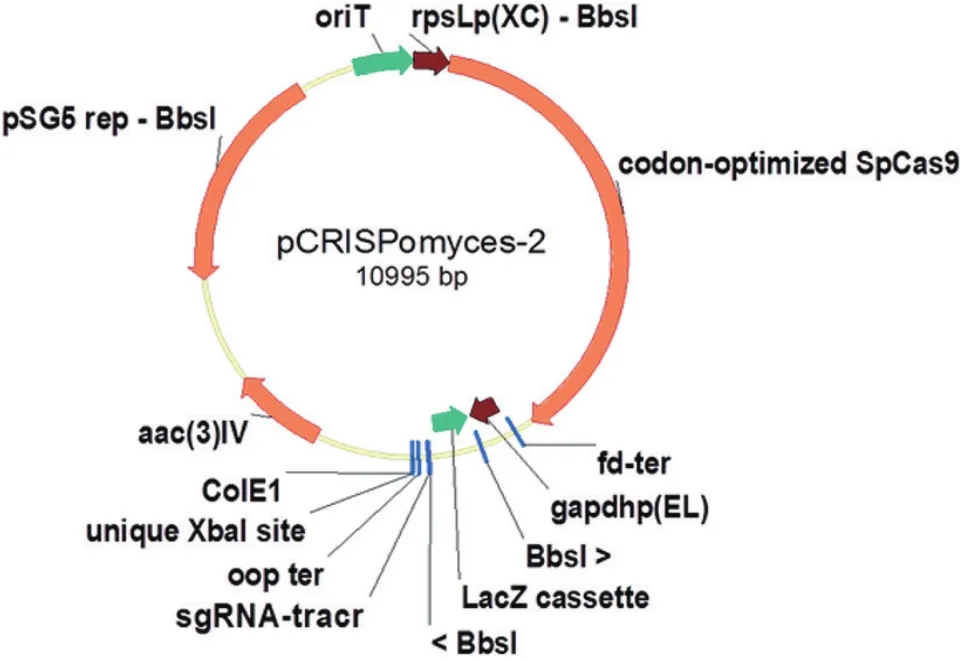
图1 pCRISPomyces-2的质粒图谱[15]
内生链霉菌SAT1分离自药用植物荠苨(Adenophora trachelioides)的根部,对16S rRNA基因进行分析发现其与阿南德氏链霉菌(Streptomyces anandii JCM 4720T)亲缘关系最近[16]。该菌的菌体和发酵液对栗疫菌(C. parasitica)有很强的抑制作用,为了深入研究其抑菌机制和代谢调控过程,实验室前期通过Pacbio sequel测序平台对SAT1进行了全基因组测序。通过antiSMASH bacterial version(http://antismash.secondarymetabolites.org/) 分 析SAT1基因组,共预测出14种类型37个次级代谢基因簇。其中hyBl基因簇与潮霉素B(已报道对真菌有抑制活性[17])基因簇有52%的同源性,可能指导潮霉素B类抗生素的合成,并在抑制栗疫病方面发挥主要作用。本文通过CRISPR/Cas9技术建立内生链霉菌SAT1的基因簇编辑方法,验证hyBl基因簇表达产物的抑菌功能,同时也为深入研究其他基因簇功能和代谢调控途径提供技术和方法。
1 材料与方法
1.1 材料
1.1.1 菌株、质粒、试剂及引物 本实验所用的菌株和质粒见表1。安普霉素、氯霉素、卡那霉素和萘啶酮酸均购自北京拜尔迪生物科技有限公司;各种限制性内切酶、T4 DNA连接酶、Taq DNA聚合酶、DNA Marker、胶回收和质粒提取试剂盒均购于大连宝生物有限公司(TaKaRa);实验中用到的引物(表2)的合成及测序由上海生工生物工程股份有限公司完成。

表1 实验中涉及的质粒和菌株

表2 实验中所用引物
1.1.2 培养基 常用的LB、2×YT、PDA、ISP2和TSB培养基配方见文献[18]。接合用MS培养基:豆饼粉20 g,甘露醇20 g,琼脂20 g,蒸馏水1 000 mL,pH自然。
1.2 方法
1.2.1 Protospacer的筛选和sgRNA cassettes的合成 作为宿主被CRISPR/Cas9系统识别切割的标记,利用 Strawberry Perl(http://strawberryperl.com/)从目标序列中筛选合适的protospacer(20 bp+NGG,共23 bp序列),特异性序列的筛选依据以下三个原则[19]:(1)优先选择 protospacer的 3'端最后四个碱基为嘌呤的序列;(2)优先选择在非编码链上的序列;(3)保证protospacer的3'端最后12个碱基和PAM组成的15 bp序列在基因组上的唯一性。根据以上原则确定最优的protospacer序列,并与gRNAtail、T7term、gapdhp(EL)及 Bbs Ⅰ 酶切位点序列共同组装成sgRNA表达盒。sgRNA表达盒序列由上海生工生物工程股份有限公司合成,并保存于质粒pUC57上(载体命名为pUC57-gRNA)。
1.2.2 CRISPR/Cas9 敲除载体的构建 载体构建过程如图2所示。首先用Bbs I分别酶切pUC57-gRNA和pCRISPomyces-2,切胶回收sgRNA cassettes和线性的pCRISPomyces-2,后者进行去磷酸化处理,然后采用Golden Gate Program(37℃ 10 min,16℃ 10 min,10个循环;50℃ 5 min,65℃ 20 min,4℃保存)将sgRNA cassettes插入pCRISPomyces-2载体。反应体系为:去磷酸化的pCRISPomyces-2,6 μL;sgRNA cassettes,10 μL ;10×T4 Ligase Buffer,2 μL ;T4 Ligase,1 μL ;Bbs I,1 μL。以 SAT1 基因组DNA为模板,利用两对overlap PCR引物对(表1,CH647_kl_s和 CH647_kl_r;CH660_kr_s和 CH660_kr_r)分别扩增两段同源臂序列,然后利用引物CH647_kl_s和CH660_kr_r进行Touchdown PCR实现两段同源臂无缝连接。反应程序为95℃ 10 min;94℃ 30 s,62℃ 30 s(每个循环下降0.5℃),72℃2 min,30 个循环 ;94℃ 10 min,47℃ 30 s,72℃ 2 min,10个循环;72℃ 10 min,4℃保存。最后通过酶切、连接,将上下游同源臂插入pCRISPomyces_gRNA载体的Xba I位点。
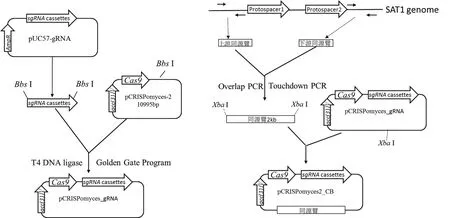
图2 敲除载体的构建
1.2.3 SAT1与E. coli的属间接合转化 取5 mL含有敲除质粒的细菌菌液(OD600=0.4-0.6)于10 mL试管中,用等体积不含抗生素的LB培养基洗涤两次弃上清,加入0.5 mL LB重悬作为供体菌。用2×YT培养基制备SAT1孢子悬液(108个/mL),50℃热激10 min后,于三角瓶中震荡孵育至孢子出芽,作为受体菌[20]。将制备好的0.5 mL供体菌和等体积孢子悬液混合,离心后弃上清用剩余液体重悬,滴加到接合培养基上,30℃共培养14-20 h。用25 μg/mL的安普霉素和萘啶酮酸各1 mL覆盖接合平板,并将接合菌饼涂布均匀,30℃恒温培养7 d,平板上出现的单菌落可初步确定为接合子。
1.2.4 突变株的鉴定及抑菌活性分析 在目标序列上设计3条引物,引物1位于上游同源臂之前,引物2位于下游同源臂之后,引物3位于hyBl基因簇上。以接合子基因组DNA为模板,分别采用引物对1和2,1和3进行PCR扩增验证(表1)。将验证成功的突变株连续转接3次以检测其遗传稳定性。之后,将其接种于不含抗生素的ISP2平板上,37℃恒温培养3 d以去除质粒。以PDA培养基上生长良好的栗疫菌(C. parasitica)为指示菌,通过平板对峙检测突变菌株的抑菌活性。以野生型Streptomyces sp. SAT1为对照,每个样品重复3次,28℃恒温培养7 d,观察结果。
2 结果
2.1 Protospacer序列的筛选、sgRNA cassettes的合成及敲除载体的构建
本研究敲除的hyBl基因簇由16个基因组成(ctg4_647-ctg4_662),长度约15 kb,为尽可能敲除整个基因簇,我们以位于基因簇首尾的ctg4_647和ctg4_660两个基因为目标,筛选两个基因序列中所有20 bp的protospacer及3'端的NGG序列(共23 bp),最终确定特异性最高的序列是GACCTCGAAG TCGTGCAGGAAGG(ctg4_647) 和 CACGGCGAGA TCGACCAGGACGG(ctg4_660),分别记为 protosp-acer1和protospacer2, 并 按5'-acgc-protospacer1-gRNAtail-T7term-gapdhp(EL)-protospacer2-gttt-3'的顺序合成sgRNA表达盒,序列如图3-A所示。其中protospacer1由质粒上Bbs I酶切位点之前的gapdhp(EL)驱动,protospacer2由表达盒内部的gapdhp(EL)驱动。gRNAtail相当于 sgRNA-tracr,协助protospacer1发挥作用,质粒上的sgRNA-tracr与protospacer2一起形成另外一条sgRNA。
通过Golden Gate Program程序将sgRNA表达盒插入载体pCRISPomyces-2中,并转化到E. coli Top10感受态细胞。鉴于sgRNA表达盒(499 bp)和载体(约11 kb)的序列长度相差悬殊,我们设计了引物Bbs I_s和Bbs I_r进行PCR验证,结果(图3-B)表明pCRISPomyces_gRNA构建成功。选择protospacer1上游和protospacer2下游各1 kb序列作为同源臂,并通过酶切、连接的方法将无缝连接的上下游同源臂插入pCRIPomyces_gRNA载体的Xba I位点,利用引物Xba I_s和Xba I_r进行PCR验证,结果(图3-C,line2)表明载体pCRIPomyces2_CB构建成功。

图3 A:sgRNA表达盒序列;B:sgRNA cassettes插入验证;C:上下游同源臂插入验证
2.2 供体菌株类型、离子和接合时间对接合效率的影响
重组载体pCRISPomyces2-CB进入SAT1细胞后,Cas9蛋白会在protospacer1和2的位置进行特异性的DNA切割,然后细胞通过同源重组修复DNA的过程中丢掉两个protospacer中间的DNA(图4-A),从而实现基因或基因簇敲除的目的。通过电转化,我们将重组敲除载体pCRISPomyces2-CB转入接合用大肠杆菌,分别获得E. coli ET12567(pUZ8002/pCRISPomyces2-CB) 和 E. coli S17-1(pCRISPomyces2-CB)两种供体菌。利用上述两株重组菌株与SAT1的孢子进行接合转移,发现以E. coli S17-1(pCRISPomyces2-CB)作为供体菌几乎没有获得接合子,而以甲基化缺陷型的E. coli ET12567(pUZ8002/pCRISPomyces2-CB)作为供体成功得到了接合子。
一定浓度的Ca2+和Mg2+可以增加接合效率,本研究分别利用含有10 mmol Ca2+和Mg2+的MS培养基作为接合培养基,结果(图5)表明,含Mg2+的培养基接合效率略高于含Ca2+的培养基。同时,接合时间对接合效率也有明显的影响。本实验条件下,16-18 h接合效果最好(图5),低于16 h,转移到链霉菌中的抗生素耐受基因未充分表达,容易被杀死;高于18 h,菌丝体生长旺盛,不易筛选到单菌落。经过以上条件的探索,我们成功获得一株突变株。如图4-B所示,利用引物对1和2可以扩增出2.5 kb的产物(line3);利用引物对1和3扩增不出任何产物(line4),与预测突变株的结果相符,初步确定该菌为hyBl基因簇缺失菌株。测序结果也表明约14 kb的片段被成功敲除。将突变株接种到不含抗生素的ISP2平板上,置于37℃培养2-3 d以去除质粒。再次接种到含抗生素的培养基上,突变株不再生长,成功得到去除了质粒的突变株,将其命名为 SAT1△hyBl。
2.3 SAT1△hyBl抑菌活性分析
连续转接3次后,SAT1△hyBl菌株形态比较稳定,且再次提基因组验证的PCR结果与上述一致,证明突变株能够稳定遗传。用ISP2固体培养基培养时,突变株的生长速度明显慢于野生株,去除质粒的SAT1△hyBl生长速度略慢于野生株(图6-a、b和c)。抑菌活性分析结果(图6-A、B和C)表明无论抑菌圈的宽度还是面积,突变株SAT1△hyBl对栗疫菌的抑制活性明显小于野生型菌株。对两者进行测量,发现野生型和突变型菌株对栗疫菌的抑菌圈宽度分别为1.17±0.047 cm和0.27±0.047 cm(表3),突变型菌株相对于野生型菌株降低了77%。综上所述,hyBl基因簇在拮抗菌SAT1抑制栗疫菌的过程中起着关键作用。
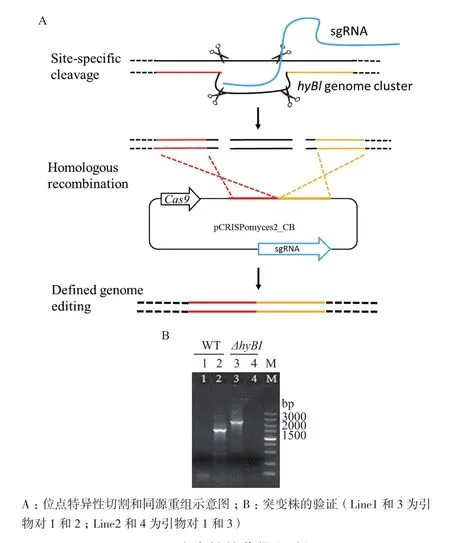
图4 突变株的获得和验证
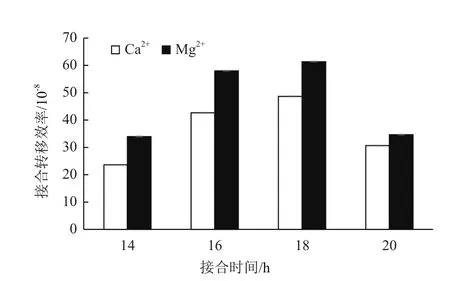
图5 不同时间和离子的选择对接合效率的影响
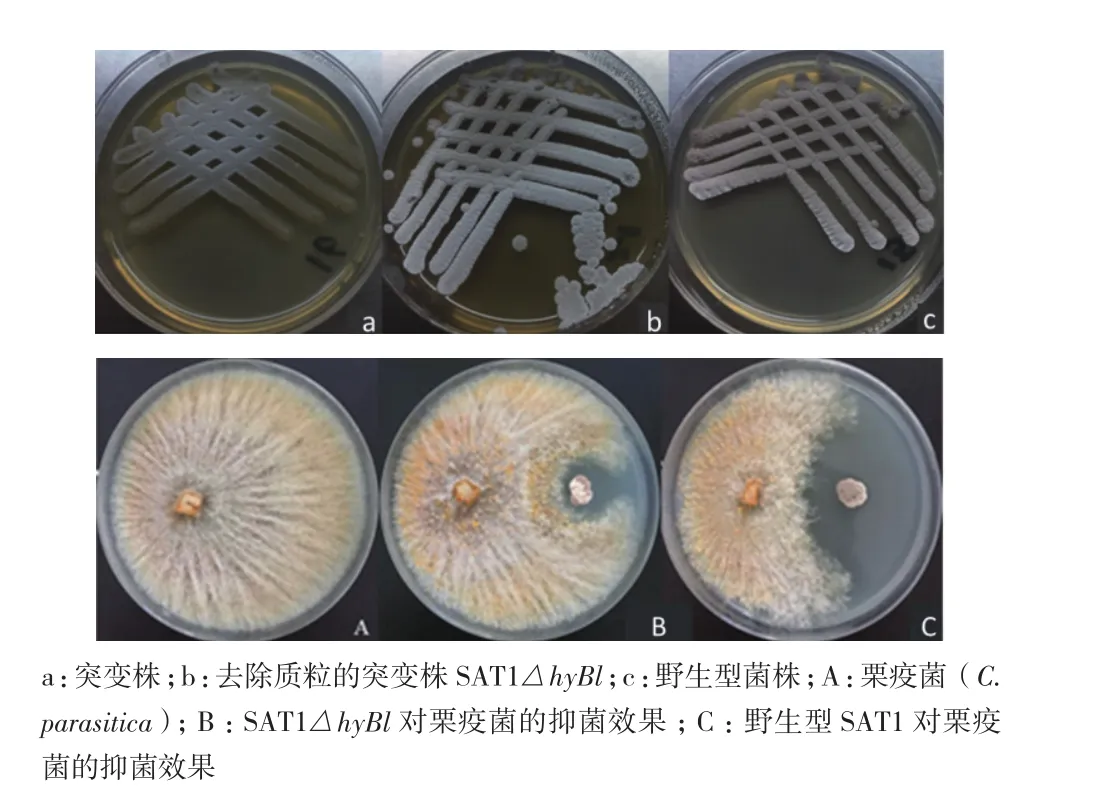
图6 野生型和突变型生长情况和抑菌活性的比较

表3 野生型和突变型抑菌圈宽度比较
3 讨论
CRISPR/Cas9系统来源于细菌和古细菌的天然免疫系统。与常规遗传操作技术相比,该系统应用于链霉菌具有明显的优势。一方面它可以任意编辑小到单核苷酸,大到几十kb的基因片段,也可以避免 Cre/loxP系统[21]、Flp/FRT系统[22]等无痕敲除方法带来的染色体不稳定,敲除周期长等缺陷,还能避免传统有痕敲除方法中筛选标记不能二次利用的缺点。利用pCRISPomyces-2质粒完成基因编辑后,可以在37℃培养去除突变株中的质粒,进而再次利用相同的筛选标记进行双基因或多基因敲除。目前,CRISPR/Cas9系统既可以用于链霉菌中代谢基因簇的功能验证、代谢调控机制的探索、干扰链霉菌中代谢旁路提高目的产物的产量,也可用于对PKS(聚酮合成酶)或NRPS(非核糖体合成多肽)模块进行局部替换以获得新的天然产物[23]。
CRISPR/Cas9编辑系统有许多优点,但仍然存在一个不可避免的问题,即对基因组其他位点非特异性切割造成的脱靶效应(Off-target effects)[24-25]。增加该系统识别位点的特异性、解决其脱靶效应是应用该技术的前提。本研究按照Cobb等的方法筛选了protospacer,并设计了同源臂以提高其特异性,从而精确地实现了对目的基因的编辑。但是属间接合转化的效率仍然较低,因此还需进一步优化SAT1的接合转移条件。目前与SAT1亲缘关系最近的阿南德氏链霉菌尚没有属间接合的相关报道,本研究也为其接合转化条件提供了参考。
构建工程菌株提高抗生素产量,发现新的抗生素对抗不断出现的多耐药细菌是当前次级代谢基因簇研究的意义所在。自Cobb等首次将CRISPR/Cas9系统应用于链霉菌以来,利用pCRISPomyces-2敲除载体的研究仍然较少。Qin等[26]通过该技术对Streptomyces formicae中的一个二型聚酮合酶基因簇(BGC30)进行功能验证,并进一步验证了该基因簇中forV基因控制卤素原子的引进这一功能。在本实验中,突变株SAT1△hyBl对栗疫菌的抑菌活性发生了显著降低,表明hyBl基因簇在抑制栗疫菌过程中发挥关键作用。但我们也发现突变株的活性并未完全消失,因此推测可能存在其他的基因簇指导合成抑制真菌的活性物质,该推测可以利用本文建立的方法进一步深入研究。另外,hyBl基因簇与吸水链霉菌中的潮霉素B基因簇仅有52%的同源性,我们推测该物质可能为潮霉素B的结构类似物,但是具体的化学结构还需后期进行物质纯化和结构解析。
4 结论
基于CRISPR/Cas9技术,本文通过protospacer的筛选、敲除载体的构建、接合转移供体菌、辅助离子的选择及接合时间等条件的探索,最终以ET12567(pUZ8002/pCRISPomyces2-CB)为供体菌,以含有Mg2+的MS为接合培养基,接合16-18 h,成功敲除14 kb的hyBl基因簇,建立了SAT1的基因簇编辑方法。突变株对栗疫菌的抑制带宽降低了77%,这说明该基因簇合成的次级代谢产物在抑制栗疫菌方面发挥重要作用。本研究为阿南德氏链霉菌(S. anandii)的基因工程操作和内生链霉菌SAT1抑菌机制的研究提供了良好的技术支撑,也从侧面验证了该系统在链霉属中应用的广泛性。
猜你喜欢
中国计量大学学报(2022年2期)2022-07-18
当代水产(2022年1期)2022-04-26
江西农业学报(2021年4期)2021-04-20
水生生物学报(2021年1期)2021-02-04
食品科学(2020年4期)2020-03-11
肿瘤防治研究(2019年7期)2019-01-06
中国钱币(2015年6期)2015-11-18
少儿科学周刊·少年版(2015年3期)2015-07-07
少儿科学周刊·少年版(2015年3期)2015-07-07
中国当代医药(2015年9期)2015-03-01
