
不同大豆连作年限对黑土细菌群落结构的影响
2019-08-20 10:57刘株秀刘俊杰徐艳霞王光华
生态学报 2019年12期
刘株秀,刘俊杰,徐艳霞,张 武,米 刚,姚 钦,王光华
1 中国科学院东北地理与农业生态研究所 黑土农业生态重点实验室, 哈尔滨 150081 2 中国科学院大学, 北京 100049 3 黑龙江省畜牧研究所, 齐齐哈尔 161005 4 黑龙江省农业科学院黑河分院, 黑河 164300
东北黑土是我国重要的土壤资源,在保障国家粮食安全和生态安全上具有重要地位。受耕地面积、经济效益和气候特征等因素影响,大豆连作在黑龙江省北部地区普遍存在。Liu等[1]对东北地区大豆连作产量统计分析表明,大豆连作4年后不仅导致产量降低40%,而且土壤的生物活性显著退化。已有研究证明,大豆连作会导致土壤理化性质改变、土壤微生物群落结构变化、土壤酶活性降低和根系分泌物的化感自毒作用加剧[2- 5]。在这些限制因子中,生物因素被普遍认为是导致连作障碍发生的主要影响因子[6]。
土壤微生物是土壤的重要组成部分,在调节土壤生态系统功能,如养分循环、有机质分解、土壤结构维持、温室气体产生和环境污染物净化起着重要作用[7-9]。已有研究证实,土壤微生物的数量、种类和多样性是维持土壤健康和质量的重要因素[10]。因此,研究不同种植制度下土壤微生物群落结构的演替规律,对选择合理种植措施和改善土壤生态功能具有重要意义。
目前关于不同种植措施下,微生物群落结构组成及多样性变化关系研究结果存在差异。如Tang等[11]采用克隆文库方法,对大豆连作和轮作细菌群落结构研究表明,大豆轮作下土壤放线菌数量和多样性指数显著提高,而其他细菌门类则呈现下降趋势。Li等[12]研究发现,在非根际土壤中大豆连作没有改变细菌群落结构,而在根际土中细菌群落结构受连作和轮作影响显著。利用高通量测序技术,Xuan等[13]研究表明在水稻-玉米和水稻-绿豆的轮作体系中,细菌数量和丰富度显著高于水稻连作处理。然而,一些研究结果显示轮作条件下,细菌群落结构和多样性指数并没有发生显著变化[14-15]。他们认为不同的研究方法(DNA指纹图谱和高通量测序)、不同的轮作体系和种植年限是导致结果不同的主要原因。
针对黑土区大豆种植面积不断增加所导致连作障碍的问题,以往关于连作下微生物群落结构演替的研究主要是采用分离培养、变性梯度凝胶电泳(DGGE)或磷脂脂肪酸(PLFA)等较低通量的研究手段,在一定程度上限制了对连作障碍下微生物群落结构变化的深入理解[16-17]。基于此,本研究采用Illumina高通量测序对黑龙江省北部黑河地区大豆不同连作年限(3、5和13年)和大豆-玉米轮作体系下土壤细菌群落结构组成和多样性分布特征进行研究,旨在揭示连作年限和轮作方式下细菌群落结构组成及多样性的异同关系。同时采用典范对应分析(Canonical Correspondence Analysis,CCA)和方差分解分析(Variance Partitioning Analysis,VPA)方法,阐明大豆连作细菌群落结构发生变化的主要环境驱动因子。
1 材料与方法
1.1 试验地概况
本试验地位于黑龙江省农科院黑河分院(50°15′12″N,127°27′40″E),试验地点属中温带大陆性季风气候,年平均气温-1.5℃,年平均降雨量为450—600 mm,3个连作小区面积分别为0.5 hm2,大豆-玉米轮作的试验区面积为1 hm2,土壤类型为黑土。供试样地长期土壤施肥和管理方式一致(每公顷化肥用量为:磷酸二胺100 kg、尿素4 kg和磷酸二氢钾83 kg)。
1.2 试验设计
本研究选取大豆连作3年处理(CC3)、大豆连作5年处理(CC5)、大豆连作13年处理(CC13)和大豆-玉米轮作5年处理(CR5)为供试土壤。于2015年7月7日在大豆开花期采集大豆非根际土壤。在试验区内采用随机采样方法,采集0—20 cm耕层土壤,每个样点随机采集10个土壤样品作为一个重复,每个处理4次重复。土壤样品去除枯枝、树根及石块等杂物后过2 mm筛,放入带冰袋的保温箱中,带回实验室4℃保存,其中部分样品保存至-80℃冰箱中用于提取土壤微生物总DNA。
1.3 试验方法
1.3.1 土壤理化性质测定
土壤pH值采用pH计测定振荡30 min后的土壤水悬浮液(1∶2.5 w/v)。土壤全碳(TC)和全氮(TN)用元素分析仪(VarioELIII,Germany)测定。土壤全磷(TP)、速效磷(AP)利用连续流动分析系统(SKALAR SAN++,Netherlands)进行测定[18]。土壤全钾(TK)和速效钾(AK)利用火焰光度计(ICPS- 7500,Shimadzu,Japan)进行测定。此外,利用碱性水解扩散法测定土壤速效氮(AN)[19]。
1.3.2 DNA提取
采用Fast DNA®Spin Kit for Soil(MP Biomedicals, USA),按照说明书进行土壤微生物总DNA的提取。提取后的DNA用TE缓冲液(10 mmol/L Tris-HCl, 1 mmol/L EDTA, pH 8.0)溶解,并用NanoDrop® 2000(Thermo Scientific, USA)测定DNA含量及质量,土壤DNA最后保存于-20℃冰箱中备用。
1.3.3 高通量测序
以提取的土壤微生物总DNA为模板,利用通用引物515F和907R[20]对细菌16S rRNA基因进行PCR扩增。25 μL的PCR体系中含有2.5 μL 10 × PCR buffer,2 μL 2.5 mmol/L dNTPs,0.25 μL Taq酶(5 U/μL)(TaKaRa, Dalian, China),0.5 μL正向和反向引物,2 μL样品DNA为模板,用灭菌超纯水补足至25 μL体系。PCR反应条件为:初变性95℃ 5 min;然后变性95℃ 1 min,复性63℃ 1 min和延伸72℃ 1 min,共30个循环;最后是延伸72℃ 5 min。每个样品做3次重复,混合后利用Agarose Gel DNA purification kit(TaKaRa, Dalian, China)进行纯化。PCR纯化后的产物送到上海美吉生物公司(Shanghai, China)利用Illumina-MiSeq平台进行双端测序分析。
1.3.4 高通量序列分析
基于Illumina PE 300测序,首先将获得细菌的FASTQ原始序列文件,通过QIIME Pipeline Version 1.8.0(http://qiime.org/tutorials/tutorial.html)[21]进行序列拆分和质量控制。利用FLASH(fast length adjustment of short reads)软件,对质控序列进行双端拼接,并去除序列长度小于200 bp,平均质量得分小于20的序列信息[18]。质控后通过Uchime algorithm[22]软件去除嵌合体序列信息。此后,通过RDP(ribosomal database project)(http://rdp.cme.msu.edu/classifier/classifier.jsp)对有效的序列信息进行分类信息注释。通过CD-HIT方法[23],基于97%的相似度水平进行OTUs分类单元划分。利用PyNAST(Python Nearest Alignment Space Termination tool)[24]软件对每个分类单元的代表性序列进行聚类分析,同时利用FastTree软件构建系统进化树[25]。
1.3.5 统计分析
本研究基于获得最小序列的CC3处理,将所有处理的序列数随机抽平至3.0万条,进行α和β多样性分析。其中Chao1指数[26]和基于系统进化距离的PD值[27],用来分析不同种植制度下细菌多样性指数的变化关系。基于SPSS(18.0版本)软件,利用One-way ANOVA,对不同处理的土壤理化性状、细菌门分类水平的相对丰度和α多样性指数进行单因素方差分析。同时,利用R(v.3.3.1)软件(R Development CoreTeam, 2006)进行聚类分析(Cluster analysis)、典范对应分析(CCA)和方差分解分析(VPA),以解析不同种植制度下,细菌群落结构组成的变化特征和主要驱动因子。
2 结果与分析
2.1 不同处理土壤的理化性质
不同处理的土壤理化性质如表1所示。与连作处理相比,除全碳(TC)和全钾(TK)含量外,大豆-玉米轮作处理(CR5)显著提高了土壤pH、全氮(TN)、全磷(TP)含量和速效养分(AP、AK和AN)含量。此外,对比不同连作年限大豆的土壤理化性状发现,除速效磷(AP)含量在连作3年处理(CC3)达到最高值外,大豆连作13年处理(CC13)的TC、TN和速效养分AN、AK含量均显著高于连作3年(CC3)和5年(CC5)处理。

表1 不同处理对土壤理化性状的影响
(1) CC3: 连作3年,continuous cropping 3 years; CC5: 连作5年,continuous cropping 5 years; CC13:连作13年,continuous cropping 13 years; CR5:轮作5年,cropping rotation 5 years; (2) 用 Duncan法统计(n=4), 同行数据后不同字母表示差异显著(P<0.05)
2.2 细菌门水平相对丰度

图1 不同种植制度下土壤细菌门水平下群落结构组成Fig.1 Composition of soil bacterial community at phylum level with different cropping systemsCC3: 连作3年,continuous cropping 3 years, CC3- 1, CC3- 2, CC3- 3, CC3- 4为4个重复;CC5: 连作5年,continuous cropping 5 years, CC5- 1, CC5- 2, CC5- 3, CC5- 4为4个重复; CC13:连作13年,continuous cropping 13 years, CC13- 1, CC3- 2, CC13- 3, CC13- 4为4个重复; CR5:轮作5年,cropping rotation 5 years, CR5- 1, CR5- 2, CR5- 3, CR5- 4为4个重复
本研究共获得细菌序列618029条(30087—44615),以97%的相似水平进行OTU聚类,共获得2696个OTU,分布在37个细菌门(图1),其中酸杆菌门(Acidobacteria)(20.13%)、放线菌门(Actinobacteria)(17.15%)、α-变形菌门(Alphaproteobacteria)(13.55%)、β-变形菌门(Betaproteobacteria)(11.74%)、绿弯菌门(Chloroflexi)(8.69%)和芽单胞菌门(Gemmatimonadetes)(7.61%)为土壤中的优势菌门(相对丰度均大于5%),占获得总细菌序列量的78.87%。
对比处理间细菌门水平相对丰度关系发现,上述优势菌门中酸杆菌门和放线菌门各处理间差异不显著;α-变形菌门在CC13处理中的相对丰度显著高于处理CC5和CR5,而β-变形菌门在CC13处理中含量最低,与CC3、CC5和CR5对比,分别降低16.24%、18.45%和4.36%。此外,绿弯菌门和芽单胞菌门在轮作CR5处理中的丰度显著高于其他连作处理。
不同处理中检测到丰度较低的一些细菌门类,如浮霉菌门(Planctomycetes)、δ-变形菌门(Deltaproteobacteria)、拟杆菌门(Bacteroidetes)、γ-变形菌门(Gammaproteobacteria)、厚壁菌门(Firmicutes)和装甲菌门(Armatimonadetes),其相对丰度变化范围在1%—5%之间。其中拟杆菌门和δ-变形菌门在轮作处理CR5中,相对丰度显著高于连作处理;厚壁菌门和装甲菌门的相对丰度在CC3和CC5处理中显著高于CC13和CR5处理;而浮霉菌门在4个耕作处理中分布较稳定,处理间差异不显著。此外,本研究检测到包括蓝细菌(Cyanobacteria)在内的,24个丰度很低的痕量菌门以及未分类的细菌门。

图2 不同种植制度下部分细菌属水平下群落结构组成Fig.2 Composition of partial bacterial community at genus level with different cropping systems
此外,研究共检测到552个不同细菌属,其中43.25%的序列注释经RDP注释后没有明确的分类信息(unclassified)。共有49和140个细菌属的相对丰度分别大于0.5%和0.1%,其中Subgroup_1的相对丰度最高,达到6.84±0.78,其次为Gemmatimonas、Gaiellales、Solibacter和SC-I- 84相对丰度分别为3.88±0.30、3.66±0.52、3.56±0.08和3.42±0.11。对比处理间细菌属水平相对丰度关系发现,上述优势菌属中Solibacter和SC-I- 84各处理间差异不显著;CR5处理显著降低了Subgroup_1 和Gaiellales的相对丰度,而显著增加了Gemmatimonas的相对丰度。此外,研究发现连作处理CC13和轮作处理CR5分别显著增加了Spingomonas(丰度>1.5%)和Nitrospira(丰度>0.5%)的相对丰度(图2)。
2.3 Alpha多样性分析
不同耕作处理的Alpha多样性指数变化关系如表2所示。各处理的覆盖率均大于98%,说明本研究获得的细菌序列覆盖度较好,其测序深度可以满足细菌群落结构组成及多样性分析。本研究将各处理间序列随机抽平到3.0万条,用以比较分析不同处理Alpha多样性大小。总体可以看出,处理CC13和CR5的Alpha多样性指数,包括获得的OTUs数量、PD值、Chao1指数和Shannon指数均显著高于处理CC3和CC5,而长期连作处理CC13与轮作处理CR5间差异不显著。该结果表明,短期大豆连作处理降低了土壤细菌多样性指数,但长期大豆连作土壤的Alpha多样性指数有恢复的趋势。

表2 不同处理对土壤中微生物群落多样性的影响
(1) OTU: 可操作分类单元,operational taxonomic units; PD: 系统发育多样性,phylogenetic diversity; (2) 用 Duncan法统计(n=4), 同行数据后不同字母表示差异显著(P<0.05)
2.4 细菌群落结构分析

图3 不同种植制度下大豆土壤细菌群落的聚类分析 Fig.3 Cluster analysis of soybean soil bacterial community under different cropping systems
基于Bray-curtis距离细菌群落结构变化的聚类分析图谱如图3所示。细菌群落结构主要划分为轮作处理(I群)和连作处理(II群)两大集群,说明连作与轮作处理间细菌群落结构差异显著。此外,在连作处理的分布集群中,区别于CC13处理(亚类 II),短期连作的CC3和CC5处理独立分布于另一个亚类中(亚类 I),说明随着连作年限的进一步延长,土壤细菌群落结构的分异度增加。
利用典范对应分析发现,土壤pH、TN、TP、AN、AP和AK是细菌群落结构发生变化的主要驱动因子(P<0.05),且所有相关因子均与大豆-玉米轮作处理呈正相关分布关系,其中土壤pH对细菌群落结构的分异贡献度最大(图4)。此外,方差分解分析(图4)发现上述土壤因子对不同种植制度引起细菌群落分异的贡献率为77.76%,其中pH对土壤微生物群落变化的贡献率最大,为8.49%,而土壤理化因子TN、TP和速效养分含量对细菌群落分异的共同解释率为32.06%。
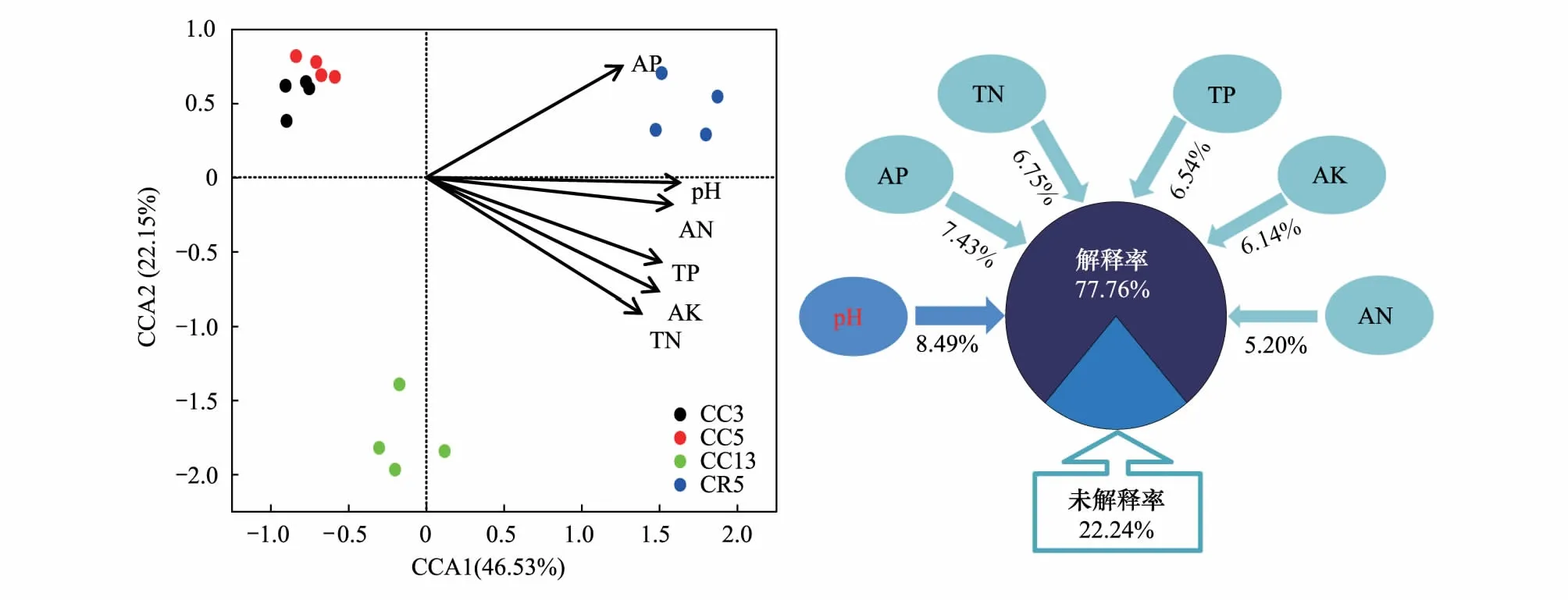
图4 不同种植制度下土壤细菌群落结构与环境因子间的典范对应分析和方差分解分析Fig.4 Canonical correspondence analysis and variance partitioning analysis between soil bacterial community structure and environmental factors under different cropping systemsCCA:典范对应分析,canonical correspondence analysis; TN:全氮,total N; TP:全磷,total P; AN:速效氮,available N; AP:速效磷,available P; AK:速效钾,available K
3 讨论
连作导致土壤理化性质恶化,养分失调和作物产量降低被广泛报道[28-30]。本研究对比轮作和连作大豆土壤理化性质发现,轮作大豆的土壤pH、TN、TP和速效养分含量(AP、AN和AK)均显著高于连作大豆。这与傅慧兰等[31]和李玉洁等[32]的研究结果相一致。轮作大豆土壤速效养分高于连作大豆,可能与轮作体系中种植玉米有关,玉米种植施肥量大,玉米茬口的残余肥料可导致后茬大豆土壤速效养分增加。与不同连作年限大豆茬口相比,轮作处理下土壤TC含量没有显著变化,该结果与Martens[33]报道的TC含量在轮作处理中高于连作处理的结果相悖。导致这种差异的原因可能是由于本研究中大豆-玉米的轮作体系只有5年,其中玉米茬口2年,尽管玉米生物量大,玉米残茬还田量大于大豆茬,但短短2年的玉米残茬还田不足以导致土壤TC产生显著的变化[34-35]。此外,对比不同连作年限处理间的土壤理化性状变化发现,CC13的土壤pH、TC、TN、TP和速效养分AN和AK含量均显著高于CC3和CC5处理,说明大豆长期连作在一定程度上改善了土壤环境,提高了土壤养分含量。王娟英等[36]对怀牛膝连作的研究发现,随连作年限的不断延长,土壤中革兰氏阳性与阴性细菌比值的降低和一些参与土壤物质循环和木质素降解的微生物类群数量的提高是导致长期连作土壤养分有效化增强和含量提高的主要原因。
土壤微生物多样性指数被认为是评价土壤生态功能的重要指标[37]。有研究证明轮作体系通过增加作物的种类和生物量的方式增加了土壤微生物的食物来源,进而导致土壤微生物的多样性升高[38]。然而,目前基于不同轮作方式下微生物多样性变化的研究结果不尽相同[39-40]。最近,Venter等[41]对不同种植方式下土壤微生物多样性指数变化研究的文章进行综合分析发现,轮作体系中仅有15.1%和3.4%的微生物群落丰富度和多样性指数呈增加的趋势。本研究发现,大豆-玉米轮作体系下,细菌群落alpha多样性指数显著高于短期连作处理CC3和CC5,而与长期连作处理CC13差异不显著。此外,本研究发现细菌群落的多样性指数与土壤pH、TP、AK、AP和AN呈显著正相关关系(P<0.001)。然而目前基于不同种植制度下,微生物群落多样性指数与环境因子间的相关性研究较少[42],Zak等[43]研究证实轮作下细菌多样性指数的增加不仅是由于作为微生物外源植物残体种类数量的增加,而土壤养分尤其是速效养分的升高是微生物多样性增加的另一主要因素。朱琳等[44]对不同大豆连作年限的研究发现,大豆长期连作(10年以上)处理增加了土壤微生物群落的丰富度和多样性。此外,本研究基于大豆长期连作CC13处理下,土壤养分含量和多样性指数的升高,间接佐证了关于土壤养分的提高导致微生物多样性升高的研究结果。
近年来,土壤pH在细菌群落结构形成中的重要作用得到了充分的论证[45-51]。如在细菌的大尺度空间分布研究[45-46]、不同海拔梯度研究[47-48]和一些定点长期野外试验研究[49-50]中土壤pH均被揭示出是决定细菌群落结构发生演替的主导因子。本研究发现,尽管不同耕作方式引起土壤pH的最大变异只有0.4个pH单位,基于CCA和VPA的分析结果均表明土壤pH与土壤细菌群落的相关性最高,且对细菌群落结构变化的单独贡献率高于其他任一个土壤理化因子,说明种植制度改变后土壤pH的变化是引起细菌群落结构发生分异的主要驱动因子。Degrune等[52]对不同种植制度下细菌群落结构研究也证实,虽然土壤pH只有0.3个单位的变化,但是土壤pH与细菌群落组成及多样性指数存在显著的相关性(P<0.01)。此外,CCA结果显示TN、TP和速效养分AN、AP和AK与细菌群落结构存在相关关系(P<0.01),且VPA结果得出养分含量对细菌群落结构变化的解释度达到32.06%,说明除土壤pH外,土壤养分是引起不同种植制度中土壤细菌群落结构变化的另一主要贡献因子。
Yin等[40]研究显示,与连作相比,轮作体系显著改变微生物的群落结构。然而,Jiang等[15]和Venter等[41]发现轮作条件下微生物群落并不发生改变,且不同的研究手段和轮作方式、种植年限等因素会导致研究结果产生差异。本研究基于高通量测序技术的研究结果显示,轮作和连作处理下的细菌群落结构在聚类分析图谱中分为二大类,而短期连作CC3、CC5和长期连作CC13处理聚为一个独立的亚类,说明大豆-玉米轮作和连作不同年限对细菌群落结构均有显著影响。此外,本研究发现一些细菌在门水平上的相对丰度发生了显著变化。如被普遍报道的富营养菌门中的拟杆菌门在长期连作和轮作处理中丰度最高,而寡营养菌门中的厚壁菌门在短期连作中丰度最高,说明上述菌门受种植制度影响后的丰度变化与其营养型关系保持一致。然而,其他富营养型菌门如放线菌门、β-变形菌门和γ-变形菌门的丰度没有发生显著变化,说明不同的种植制度在一定程度上改变了上述菌门的r型生长策略[34,53]。此外,研究发现具有土壤污染物降解功能的芽单胞菌属(Gemmatimonas)[54]和将土壤中的亚硝酸氧化成硝酸盐的硝化螺旋菌属(Nitrospira)在轮作处理中的相对丰度显著高于其他处理,说明轮作方式有助于降解土壤污染和提高土壤的氮肥力[55]。另外,对比三个连作处理发现,α-变形菌门中能利用苯甲酸、水杨酸等物质作为唯一碳源[56]的鞘氨醇单胞菌属(Sphingomonas)的相对丰度在连作13年处理中相对丰度最高,鞘氨醇单胞菌属能够降解土壤有毒物质,并且有助于植物抵抗病原菌[57]。Wei等[58]对连作大豆根部微生物群落结构数量变化关系研究发现,长期连作显著增加了哈茨木霉(Trichodermaharzianum)、厚垣普可尼亚菌(Pochoniachlamydosporia)和拟青霉菌(Paecilomyceslilacinus)的数量,导致长期连作土壤对大豆根部真菌病原菌和线虫产生了抑制作用。上述研究结果证明,长期大豆连作改变了土壤微生物群落结构,有助于促使抑病土的产生。此外,近年来的研究结果也发现,长期连作大豆的株高、生物量和产量均显著高于短期大豆连作[12]。
综上所述,本研究发现区别于连作处理,大豆-玉米轮作体系下的细菌群落结构在聚类图谱中被划分为独立的集团,说明轮作后细菌群落结构组成发生了明显的分异。一些细菌的相对丰度受种植制度影响发生了显著变化,其中富营养型的拟杆菌门和寡营养型的厚壁菌门相对丰度的变化与其营养型分布关系一致,而富营养型放线菌门、β-变形菌门和γ-变形菌门没有因轮作处理提高养分含量的情况下,而显著增加其相对丰度。此外,与连作处理相比,CR5处理显著提高了细菌群落的丰富度和多样性指数,说明轮作处理有助于改善土壤环境。有趣的是,本研究发现长期连作CC13处理显著提高了土壤养分含量和细菌群落的丰富度和多样性指数,说明长期大豆连作在一定程度上改善了土壤环境,提高了土壤的保肥能力,为土壤微生物提供了营养和能源物质。本研究利用高通量测序技术,提供了长期连作后细菌群落多样性升高的实例,然而关于土壤-作物-微生物三者如何介导连作退化土壤环境改善的内在机制尚需进一步研究。
猜你喜欢
当代水产(2022年8期)2022-09-20
昆明医科大学学报(2022年2期)2022-03-29
今日农业(2021年11期)2021-11-27
食品安全导刊(2021年20期)2021-08-30
河南科学(2020年3期)2020-06-02
中国比较医学杂志(2020年4期)2020-05-26
水生生物学报(2019年4期)2019-07-20
生物安全学报(2019年3期)2019-02-15
川北医学院学报(2019年6期)2019-02-10
中国化肥信息(2018年12期)2018-03-01
