
压力超负荷致小鼠心肌肥厚中miR-378对热休克转录因子-1的调节作用
2019-09-23 12:27邹云增
中国临床医学 2019年4期
苑 洁,邹云增
复旦大学附属中山医院心内科,上海市心血管病研究所,上海 200032
心血管疾病是全球范围内危害人类健康的“头号杀手”。心肌肥厚是许多心脏疾病如高血压、心瓣膜病、充血性心力衰竭及先天性心脏病的代偿反应,会导致心力衰竭。而血流动力学压力超负荷是引起心肌肥厚进而发生心力衰竭的主要因素。心脏在压力负荷初期通常表现为代偿性心肌肥厚,以满足机体的各种生命活动的需要。然而当心脏在持续压力超负荷情况下,代偿性心肌肥厚会发展为失代偿性心肌肥厚,从而诱发心功能不全和心力衰竭,增加猝死发生风险[1-2]。
热休克蛋白转录因子1(HSF1)在许多病理条件的(缺血损伤、氧化应激、压力超负荷等)刺激下均可以对心脏起保护作用,并在适应性的生理性心肌肥厚中发挥重要作用[1-5]。压力超负荷下,HSF1能促进心脏血管新生、抑制心肌纤维化、维持心肌重构的早期代偿[6-7],然而有关其调控机制的研究报道甚少。因此,研究HSF1在心肌肥厚中的调节机制,对于临床治疗中心功能的改善具有重要意义。
MicroRNA(简称miRNA)是广泛存在于真核生物中长度约为21 nt的内源性非编码RNA,参与基因的转录后调节,在生物体内的生理及病理过程中都扮演着重要角色[8-10]。已有研究证实,很多miRNAs参与心肌肥厚发生发展过程中重要基因的调控[9,11-12]。MiRNA是否也参与心脏保护因子HSF1的调控,值得进一步研究。
1 材料与方法
1.1 小鼠压力超负荷模型(TAC)的建立 C57B/L6雄性小鼠,周龄为10~12周,体质量18~22 g,购于上海中科院斯莱克实验动物有限责任公司。小鼠麻醉后卧于鼠板固定,气管插管后连接呼吸机,沿胸骨左侧的第二肋骨剪断肋骨,在主动脉凸侧分支的第一和第二分支之间将丝线穿过主动脉弓,用31G针头造成升主动脉环行缩窄,之后逐层缝合。假手术组只穿线不结扎。
1.2 小鼠心脏超声和血流动力学测定 对小鼠假手术组和TAC模型术后2周进行心功能和动脉收缩压(ASP)测定。使用加拿大VisualSonics公司Vevo 770超声诊断仪和30 MHz高频探头进行小鼠的心脏超声检测心脏功能,记录B-Mode图像,测量左室舒张期前壁厚度(LVAWd)、左室射血分数(LVEF)等指标。使用Millar导管对小鼠ASP进行测定。
1.3 心肌细胞培养 取1~3 d SD大鼠乳鼠15只,购于上海中科院斯莱克实验动物有限责任公司。用75%乙醇消毒,取心尖大部用PBS洗2次,将组织剪碎。用0.1%胰酶溶液37℃消化5~6次,每次8 min,收集上清到DMEM标准培养基中终止消化,1 200r/min 离心5 min,将细胞沉淀收集到1个装有DMEM低糖标准培养基中的收集管中,将收集得到的细胞悬液加入2个10 mm培养皿中,置于CO2培养箱进行差速贴壁法分离。2 h后,吸取上清,按照1×106/mL将细胞接种,次日换液,加入新鲜的DMEM低糖培养基。
1.4 MiR-378模拟物或抑制物细胞转染 心肌细胞贴壁24 h后,换成无血清无抗生素低糖培养基预处理过夜,使用转染试剂siPORTTMNeoFXTMTransfection Agent(Fisher, AM4510)转染miR-378 模拟物 (35 nmol/L) (Applied Biosystems,AM17100)或抑制物(50 nmol/L) (Applied Biosystems,AM17000)分子以及FAM标记模拟物和抑制物的阴性对照。48 h后收集细胞或对细胞进行机械牵张刺激。
1.5 荧光素酶报告基因检测 将扩增得到的HSF1野生型基因3′UTR或突变型片段各自插入到pMIR-REPORTTMluciferase miRNA expression(Applied Biosystems,AM4510) 报告载体中得到Luc-HSF1及种子序列突变载体Luc-HSF1-MU,使用FuGENE HD(Roche,E2311)根据分组将荧光素酶报告载体和miR-378模拟物或抑制物共转染Cos7细胞,48 h后使用Dual-Light○RSystem(Applied Biosystems,T1003)检测化学发光值。
1.6 miRNA表达水平检测 用TRIzol试剂(InvitrogenTM,15596018)从心肌组织和心肌细胞中提取总RNA,并通过NanoDrop ND-1000分光光度计测量浓度。使用TaqMan MicroRNA Assays (Applied Biosystems,4427975)在ABI 7500系统进行qRT-PCR反应,U6作为内参,实验步骤按照试剂盒操作。
1.7 Western 印迹检测蛋白表达 心肌组织或心肌细胞用RIPA(碧云天,P0013C)裂解,混匀后4℃振荡4 h,以12 000×g离心15 min(4℃),上清为总蛋白质。Western印迹按常规步骤电泳,每孔加入20~40 μg蛋白量,使用HSF1(CST,4356)、Hsp27(CST,95357)抗体和HRP偶联的兔二抗(CST,7075)检测各组蛋白表达水平,以GAPDH为内参计算其相对表达水平。

2 结 果
2.1 小鼠心肌组织内HSF1和miR-378的表达变化 小鼠心肌肥厚(图1A)代偿期时心肌HSF1表达显著升高,而miR-378的表达显著下降。血流动力学检测结果显示:TAC术后小鼠的ASP、收缩压、左心室舒张末期压力、收缩末期压力均明显升高(P<0.05),心肌肥厚处于代偿期,心功能无显著下降,LVEF值与假手术组相比无显著差异(图1B)。小鼠心脏超声数据结果显示:TAC手术2周后心肌肥厚显著,心脏质量/体质量比值HW/BW、左室壁厚度显著增加(P<0.05,图1C)。TAC小鼠手术2周后心肌内HSF1及其下游调控蛋白Hsp27的表达均显著升高(图1D),而心肌内miR-378的表达显著降低(图1E)。

图1 压力超负荷导致心肌肥厚小鼠心肌内HSF1和miR-378的表达变化
A: 小鼠TAC 术后2周及假手术组心脏外观、血流动力学变化;B:小鼠TAC术后ASP和LVEF表达变化; C:小鼠TAC术后LVAWd和HW/BW表达变化;D:Western印迹检测TAC 术后2周小鼠心肌内HSF1和Hsp27蛋白表达变化; E: 实时定量PCR检测TAC术后2周小鼠心肌内miR-378的表达变化.*P<0.05与假手术组相比;n=5
2.2 心肌细胞体外牵张刺激下HSF1和miR-378的表达变化 使用0.01%鼠尾胶原溶液按照6~10 μg/cm2的浓度包被硅胶皿,室温下在无菌操作台内将硅胶皿风干后。心肌细胞接种到硅胶皿培养后,将硅胶皿放在机械牵张板上拉伸,使得其长度可以增加20%。对体外培养的心肌细胞机械牵张刺激12 h、24 h,检测心肌细胞内HSF1和miR-378的表达变化,发现HSF1的蛋白表达水平在牵张24 h后显著升高(P<0.05,图2A),miR-378的表达水平在机械牵张24 h后却显著下降(P<0.05,图2B)。

图2 心肌细胞体外牵张刺激下HSF1和miR-378的表达变化
2.3 MiR-378对 HSF1的转录后调控 心肌细胞分别转染miR-378模拟物(Pre-miR-378)、miR-378抑制物(Anti-miR-378)及FAM标记的阴性对照组(mock),24 h后分别在普通光镜下和荧光显微镜下观察细胞,发现90%以上的细胞发出绿色荧光,转染效率达到90%(图3A)。转染48 h后,Real-time PCR结果显示,转染模拟物组心肌细胞中miR-378的表达量显著升高,转染抑制物组心肌细胞内源miR-378的表达被显著抑制(图3B)。检测HSF1蛋白表达,转染miR-378模拟物组HSF1的表达被显著抑制(P<0.05),转染miR-378抑制物组HSF1表达显著升高(P<0.05,图3C)。心肌细胞过表达miR-378能够显著抑制机械牵张引起的HSF1升高(图3D)。
分析HSF1 3′UTR区域,发现在第1743~1765个碱基区域内有miR-378的潜在靶向结合种子序列(图3E)。构建包含HSF1 3′UTR的荧光素酶报告基因载体Luc-HSF1及种子序列突变载体Luc-HSF1-MU,转染Cos7细胞。当细胞过表达miR-378时,转染Luc-HSF1组细胞内化学发光被显著抑制,而Luc-HSF1-MU组则升高(P<0.05);当Cos7单独转染Luc-HSF1时,细胞内化学发光被显著抑制,而当抑制内源miR-378时,化学发光的抑制作用被逆转(P<0.05,图3F)。因此,这些结果证实miR-378能够通过靶向结合HSF1 3′UTR调控HSF1的表达。
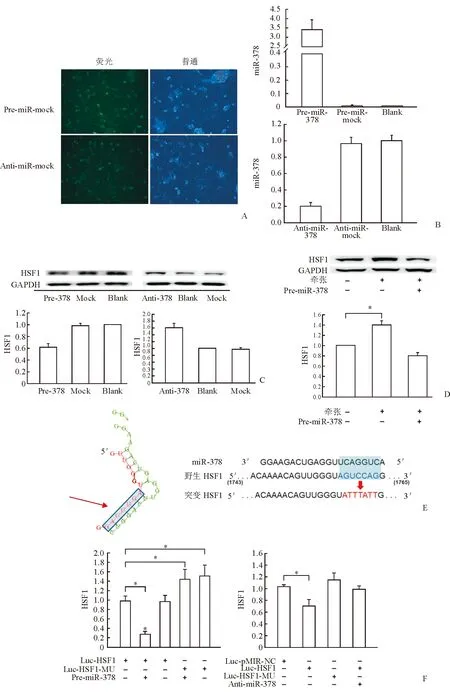
图3 MiR-378对 HSF1的转录后调控
A:心肌细胞转染FAM标记的miR-378模拟物和抑制剂(阴性对照),荧光普通光镜观察发现,转染效率达90%; B:转染miR-378模拟物和抑制物后,miR-378在心肌细胞中的表达情况; C:Western 印迹检测心肌细胞过表达和抑制miR-378后HSF1的表达变化;D:Western 印迹检测机械牵张刺激后心肌细胞内HSF1的蛋白表达;E:miR-378在HSF1 3′UTR的靶向结合位点,以及HSF1 3′UTR种子序列的突变型;F:荧光素酶保护基因检测示miR-378可直接靶向结合HSF1 3′UTR.*P<0.05,n=3
3 讨 论
HSF1在多种病理条件刺激下,如缺血损伤、氧化应激、压力超负荷等,均对心脏起到一定的保护作用[3,6,13]。另外有研究证明,当机体内的HSF1表达降低时,心脏对外界刺激的适应性会随之降低[14]。这些均表明在代偿性心肌肥厚反应中,HSF1可能是一种生理性的保护因子,但是对于其引起这种保护性效应的机制,目前研究还是十分有限。
在心肌损伤中,一些miRNA与HSF1之间的调节作用被报道。HSF1能够激活miR-135b的表达,促进内皮细胞的增殖[15];在心肌肥厚中,HSF1通过调节HSP70的表达促进miR-23表达的升高[16];在急性心梗的小鼠中,敲除内源性HSF1会促进miR-208的表达[17];房颤发生后,通过调节HSF1可引起miR-432表达的升高[18];miR-1可以上调HSF1的表达,当miR-1表达下调时会导致心肌肥厚的发生[19]。本研究通过构建心肌肥厚的代偿模型,从在体和细胞水平同时观察内源性HSF1和miR-378的表达变化,并对其中的调控机制进行了探讨。
本研究中,TAC处理2周后,小鼠表现为代偿期的病理性心肌肥厚,伴随心肌内源性HSF1及其下游调控蛋白Hsp27的代偿性升高,而心肌内源性miR-378的表达却明显下降。细胞水平发现机械牵张刺激心肌细胞会导致内源性miR-378的表达下降,而HSF1的表达则会升高。之前的研究[1-5]认为HSF1在适应性心肌肥厚中发挥重要作用,在心肌肥厚的早期代偿阶段表现为代偿性的表达升高。那么在心肌组织中,HSF1的代偿性升高是否受到miR-378的调控?本研究在心肌细胞中分别过表达和抑制miR-378,发现细胞过表达miR-378时会抑制HSF1的表达,而抑制miR-378的表达会使内源性HSF1的表达升高,并且在机械牵张刺激下,过表达miR-378会抑制HSF1的升高,证实了miR-378参与了HSF1的表达调控。利用生物信息学软件miRanda和RNAhybrid分析miR-378的潜在靶点,发现HSF1的3′UTR上有miR-378的靶向结合位点,且最小自由能为-28.4 kal/mol,说明其可以为miR-378的结合提供很好的空间结构,本研究进一步利用荧光素酶报告基因证实了miR-378可以靶向结合HSF1 3′UTR,进而调控其转录后的翻译。
综上所述,HSF1作为心肌内源重要的保护因子,在心肌缺血缺氧、机械应力及炎症损伤中均发挥了重要的保护作用。在高血压心肌肥厚的早期代偿阶段,心肌miR-378表达的下降使得其对内源性HSF1转录后抑制作用减弱,进而对HSF1的代偿性升高发挥了重要的调控作用。由于miRNA表达及调控的时序性的复杂性,有关miR-378通过调节HSF1在心肌肥厚发生发展阶段所发挥的生物学功能需要进一步深入研究,同时miR-378与miR-1是否协同调节HSF1也需要深入探讨。该研究对于HSF1参与调节心肌保护的机制提供了新的理论基础,为心肌重构干预靶点的开发提供了新思路。
猜你喜欢
临床肝胆病杂志(2022年5期)2022-11-24
今日健康(2022年3期)2022-11-21
农业工程学报(2022年12期)2022-09-09
临床肝胆病杂志(2022年8期)2022-09-07
昆明医科大学学报(2021年8期)2021-08-13
青岛大学学报(医学版)(2021年1期)2021-03-03
心肺血管病杂志(2020年5期)2021-01-14
心理学报(2020年7期)2020-07-13
医学新知(2019年4期)2020-01-02
体育科学(2018年12期)2019-01-04
