
NH3在Ir(211)和Ir(221)表面吸附的第一性原理计算
2019-12-06 09:31肖香珍
原子与分子物理学报 2019年6期
肖香珍, 杨 理
(河南科技学院 实验管理中心,新乡 453003)
1 引 言
NH3的一个非常重要的应用就是作为燃料电池中H2的来源和储存物质[1-3],其含H量远高于其它含H物种,具有很高的质量效率; NH3在通常情况下是液态,较易于储存和运输;另外,和其他的制氢方法相比,NH3能够催化氧化成无COx的H2和N2[4-6],NH3还可以用于生产含氮产物例如炸药、肥料等.另一方面,NH3也是一种重要的环境污染物,间接导致酸雨的形成,造成河流和湖泊的污染.鉴于氨气在工业和环境方面的重要性,NH3的吸附与催化分解受到研究者们的关注.理论研究相较于实验研究可以在微观层次上获得每一个基元反应的相关信息.在理论与实验上,曹益林、谢代前、Krekeberg、Goodmen等分别已经对Ir(100)、Ir(110)、Ir(111)等[7-12]表面NH3的吸附和解离进行了报道.关于NH3在Ir(211)和Ir(221)台阶面上的吸附与分解理论上目前还没有相关研究,本论文运用第一性原理密度泛函理论(DFT)计算了NH3在Ir(211)和Ir(221)面上的吸附稳定性和偏好位置以及吸附的电子结构本质.
2 计算模型和方法
优化得到Ir的晶胞参数3.88 Å与实验结果3.84 Å吻合很好[13].所有计算均采用DFT方法[14-16],单电子波函数采用平面波基组展开,截断能为400 eV,交换相关能采用GGA-PW91[17],离子芯势采用PAW方法描述[18],不可约布里渊区积分采用Monkhorst-Pack方案划分K点网格[19],Ir(211)和Ir(221)表面K点网格密度分别取为3×5×1和 5×3×1.本文采用周期性的Slab模型来模拟表面,所用表面均从优化后的结构截取,Ir(211)和Ir(221)表面均取为包含5个Ir原子层的(2×1)和(1×2)超元胞结构,吸附物放在表面一侧,顶层表面原子和吸附物驰豫且不考虑对称性,下面四层固定,选择这样的模型,既较符合实际情况,也能节省计算时间[20].层间的真空层设定为15 Å,以确保平板间的相互作用足够小.对于所有优化得到的稳定点构型通过频率分析加以验证,即局域极小点对应全部实频,吸附能的计算公式为:
Eads=Egas+Esurf-Egas/surf
其中,Egas、Esurf和Egas/surf分别表示吸附分子的总能量、清洁金属表面的总能量和吸附体系的总能量,按照此定义,吸附能Eads是将吸附体系NH3/Ir分离成没有相互作用的金属slab与NH3分子所需的能量.
3 结果与讨论
3.1 吸附能及吸附结构几何参数
Ir(211)和Ir(221)面结构相似,差别仅是台阶面大小不同,Ir(221)台阶面更宽.对于Ir(211)和Ir(221)两个表面来说,暴露出来的台阶面具有(111)面的结构特征,即每个原子周围有等距离的六个原子,两个表面结构均较(100) 、(110)和(111)面复杂,我们选择其中典型位点进行计算.对于Ir(211)面,如图1所示,top表示台阶原子的顶位,brg是台阶原子的桥位,ebrg是台阶原子与紧邻台阶面上原子形成的桥位,hcp1和hcp2是台阶面上的三重空位,二者差异是到台阶面的距离不同,fcc1和fcc2是台阶面上的三重空位fcc,两者也是到台阶面的距离不同,Ir(221)面上吸附位点的表示和Ir(211)类似.
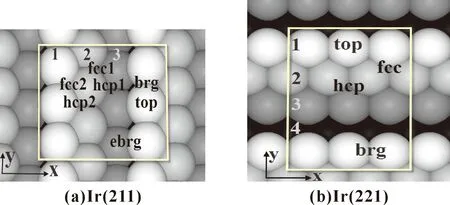
图1 NH3吸附在两个Ir面上可能的吸附位置,Ir原子由浅入深分别代表第一、二,三、四层.Fig. 1 The possible sites of NH3 adsorbed on Ir surfaces. Ir atoms represent the first, second, third and fourth layers from shallower to deeper, respectively.
对于吸附结构,主要考虑NH3在Ir(211)和Ir(221)面上的典型吸附位置,所有吸附结构中均以N原子吸附在金属表面,计算结果列于表1,NH3的最稳定吸附位均是台阶的顶位(top),这和其他一些金属的稳定吸附位置相同[21-25],说明NH3的最稳定吸附位置与金属的种类无关.吸附能都在1.0 eV以上,相差0.02 eV,均为化学吸附.由于台阶面的活性较高,NH3在这两个表面上吸附时,与在Ir (100)、Ir (111)面上NH3的吸附结构[12]有所不同,而与NH3吸附在Ir (110)面[26]类似,不是以NH3的C3轴垂直于表面的顶位.
在Ir(211)面上,NH3以N-Ir键与台阶面成99.1°角的构型,NH3的C3轴对称性减弱,吸附构型如图2,在Ir(221)面上,NH3以N-Ir键相对于台阶面成100.9°的方式稍微有些倾斜地吸附在此表面.在两个表面上,N-H键长与气相NH3分子(1.02 Å)相比均无明显变化.Ir(211)面上,NH3的三个H-N-H键之间的夹角分别为H1NH2=109.2°、H1NH3=110.2°、H2NH3=108.8°,;Ir(221)面上,三个H-N-H键之间的夹角分别为H1NH2=108.9°、H1NH3=109.4°、H2NH3=109.0°,都有所增加(相对于气相的∠HNH =107.3),结果表明,NH3分子稳定吸附两个表面后在其N-H键长基本不变的情况下,其H原子与表面更亲近些.
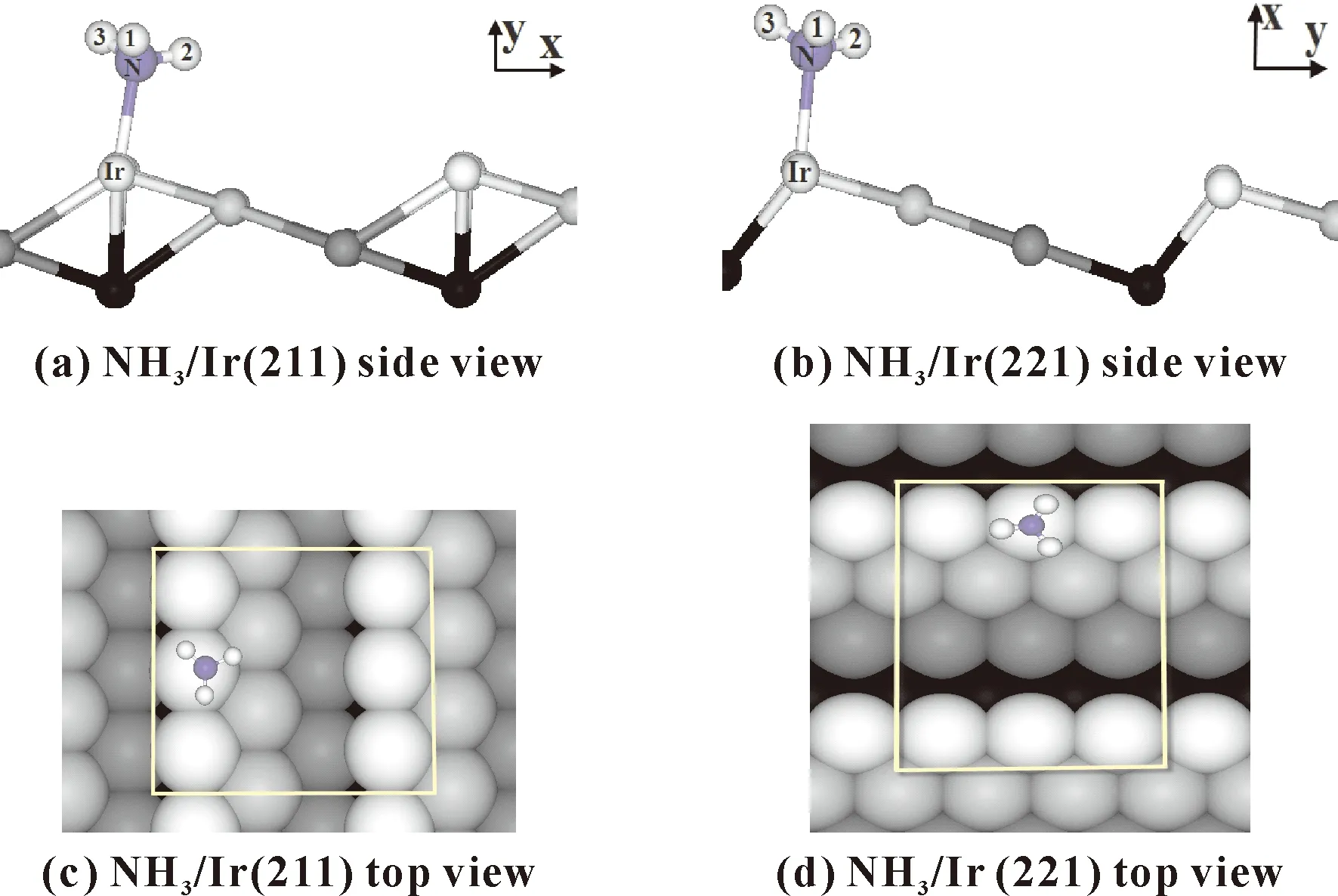
图2 NH3在Ir(211)和Ir(221))的最稳定吸附构型Fig. 2 The most stable adsorption configurations of NH3 at Ir(211) and Ir(221) surfaces
表1 NH3在Ir(211)表面最优吸附位置的吸附能和几何参数
Table 1 Adsorptionenergies and geometric parameters of NH3at the optimum adsorption location on Ir(211) and Ir(221) surfaces
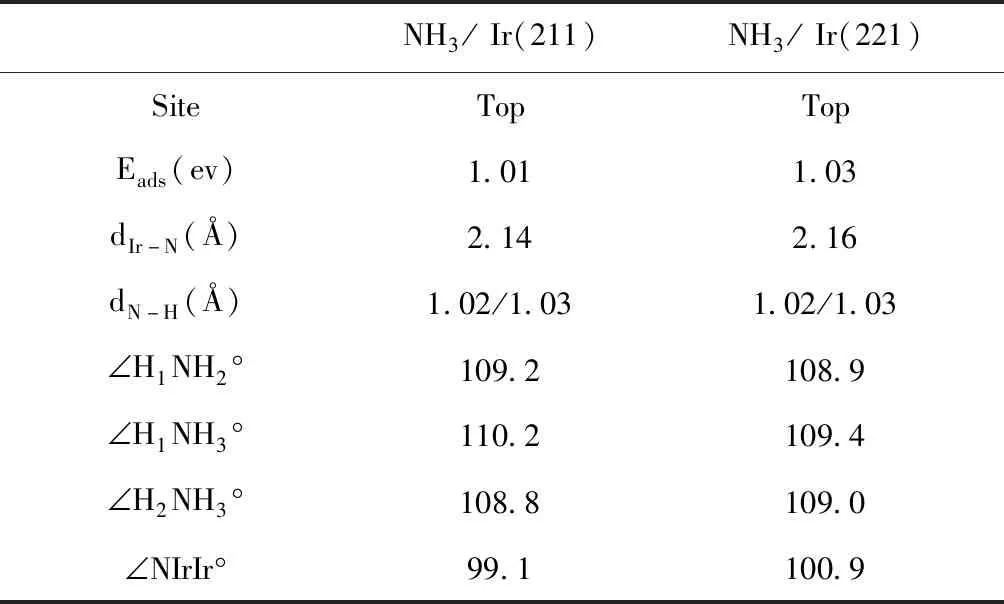
NH3/Ir(211)NH3/Ir(221)SiteTopTopEads(ev)1.011.03dIr-N(Å)2.142.16dN-H(Å)1.02/1.031.02/1.03‰H1NH2°109.2108.9‰H1NH3°110.2109.4‰H2NH3°108.8109.0‰NIrIr°99.1100.9
3.2 NH3/Ir(211)和NH3/Ir(221)的电子结构
从电子结构层次上可以解释NH3在Ir表面上的吸附成因,我们计算了气相NH3分子及两个吸附体系的电荷密度,结果示于图3.气态NH3分子的2a1、1e、3a1轨道电荷密度如图3(1)所示,可以看到NH3分子的2a1轨道上的电荷将形成分子的4个原子包围其中,1e轨道上的电荷主要堆积在N-H键上,这两个轨道是N、H形成NH3分子的主要贡献者.3a1轨道电荷主要在N原子的上、下部分,对于N-H键几乎没有贡献,由此可知,3a1轨道上电荷的变化,不会对NH3结构有明显影响,而如果1e、2a1上电荷转移的话,则NH3结构将会发生显著变化.
NH3/ Ir(211)和NH3/ Ir(221)两个体系中,由左至右分别对应于吸附后NH3分子的2a1、1e、3a1电荷密度分布,如图3(2)(3)所示.同气相NH3相比,两个体系的共同特征是,3a1轨道与表面金属Ir在垂直方向上存在明显混合,是NH3吸附的主要原因.1e和2a1轨道上的电荷与气相时基本相同,仍然定域在NH3上,即吸附没有引起NH3的1e及2a1轨道上电荷的变化,也就是说,1e和2a1轨道对NH3的吸附基本没有贡献,类似于NH3在其他体系表面的吸附成因[27].
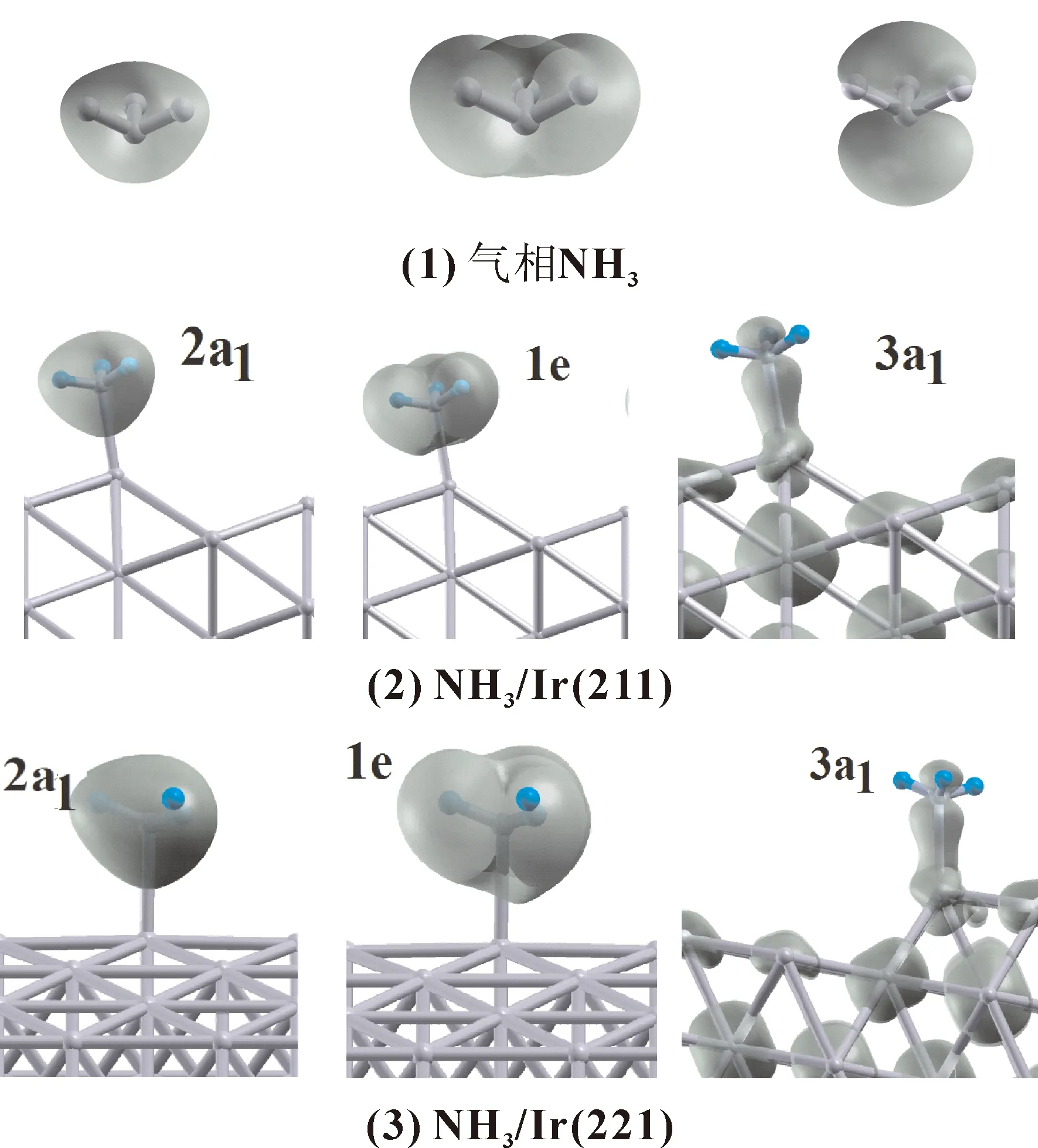
图3 气态NH3以及NH3/ Ir(211) 、NH3/ Ir(221)两个吸附体系的2a1、1e 、3a1三个轨道对应的电荷密度图Fig. 3 Charge density Maps of gaseous NH3 , NH3/Ir(211) and NH3/Ir(221) adsorptive systems for 2a1, 1e and 3a1 orbits
我们对优势吸附结构的态密度(DOS)做了进一步计算.NH3分子的价轨道按能量由低到高依次为2a1、1e和3a1.其中,2a1轨道主要由N原子的2s轨道和氢原子的1s轨道形成,是NH3分子的成键轨道,1e轨道由N原子的2px及2py轨道与氢原子的1s 轨道混合而成,3a1轨道则由N原子的2pz轨道组成,是NH3分子的最高占据轨道.当NH3吸附于表面时,3a1轨道应当起主要作用,而1e轨道作用则会比较弱,2a1轨道由于其能量更低,对NH3的吸附贡献将基本可以忽略.
从图4左边上下两图来看,最下一格是气相NH3分子的轨道能级图,倒数第二格是投影到吸附后的N原子的DOS图,图中显示有三个明显的峰,费米能级位于0 eV处.其中 2a1轨道能量较低,NH3吸附时对该轨道的影响可以忽略,将气相NH3中2a1轨道与其衍生态2a1的DOS峰对齐,在这基础上比较吸附对其它轨道的影响.1e轨道与气相NH3中相比,该态向低能方向移动了约0.1 eV,说明NH3的1e轨道与金属表面有着很弱的作用.吸附后带的3a1轨道,明显向低能量方向发生位移,可见NH3吸附时,其3a1轨道同金属表面态之间存在很强的相互作用,是NH3吸附表面的主要原因.DOS向N原子各轨道投影结果(图3左边两图的2px、2pz、2py、2S)也清楚地表明,3a1轨道同金属Ir表面态之间相互作用实际上是N原子的2pz轨道同金属Ir在Z方向上的混合,N原子的其它轨道则几乎不参与NH3在Ir表面上的吸附.右边上下两个DOS图为NH3在两个表面吸附后的总态密度(灰色线所示)以及与N 原子相连的金属Ir的5dz2(黑线所示)的投影态密度,可以看出,2a1、1e轨道同表面态之间几乎没有相互作用,因而对NH3的吸附贡献较小,而3a1轨道同相连的金属Ir的5dz2之间存在较强的相互作用,由这些态混合而成的衍生态向低能方向发生了位移,是NH3稳定吸附于表面的主要原因.
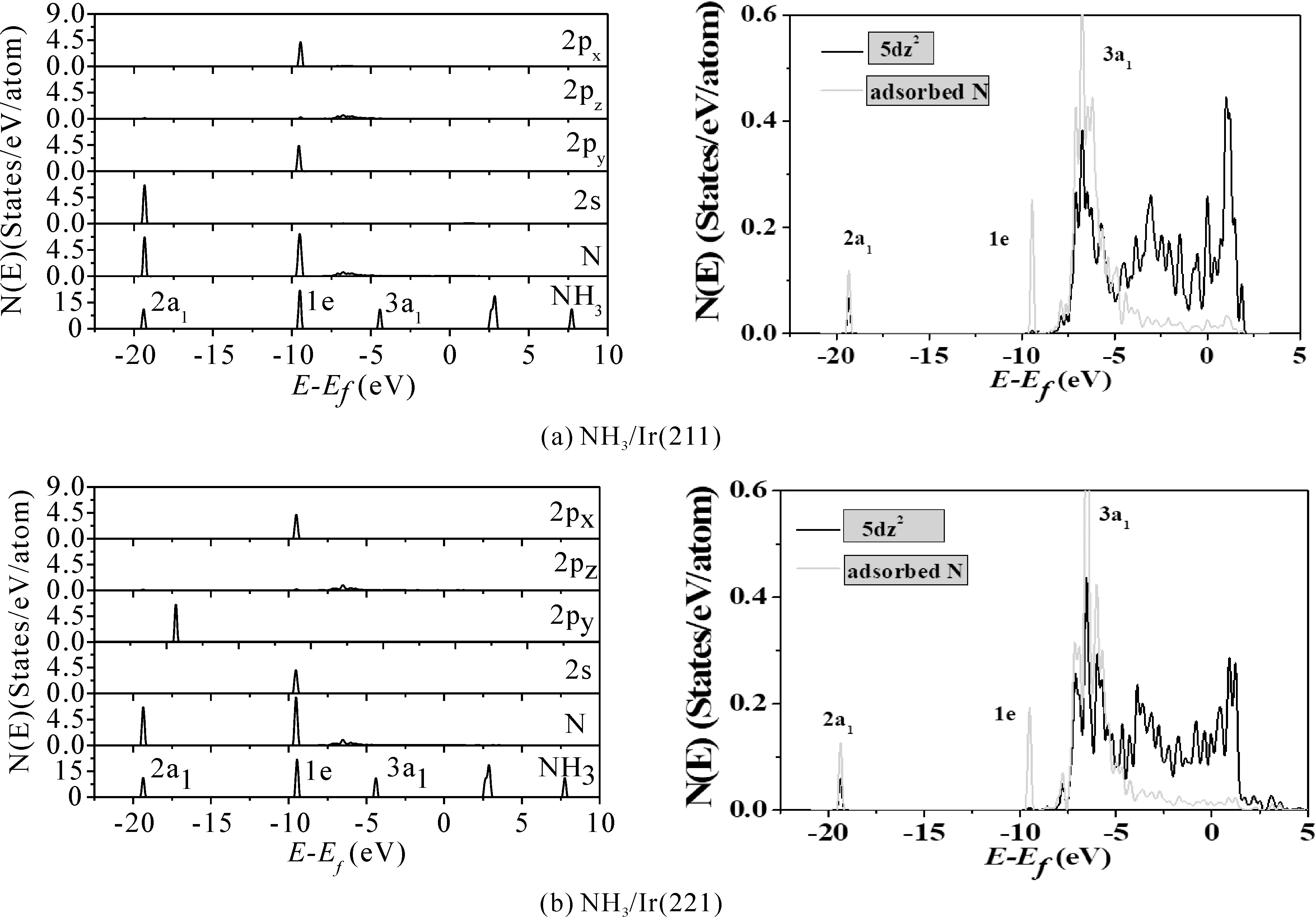
图4 两个吸附体系的态密度.将态密度投影至气相NH3、吸附后的N原子、以及N 原子的2s 、2py 、2pz和2px轨道,投影到吸附位点上Ir原子的5dz2轨道.费米能级为零点Fig. 4 The densities of states of the two adsorption systems. The density of states is projected to gaseous NH3, the 2s, 2py, 2pz and 2px orbits of N atoms after adsorption, and to the 5dz2 orbits of Ir atoms at the adsorption sites. The Fermi level is zero
4 结 论
通过对NH3/Ir(211) 、NH3/Ir(221)两个吸附体系不同位置几何结构的优化,虽然Ir(211) 、Ir (221)台阶面有些不同,但NH3的最稳定吸附位均为顶位,吸附能都在1.0 eV以上,相差0.02 eV,较为稳定,是化学吸附.电荷密度的计算结果表明两个面上,1e和2a1轨道对NH3的吸附基本没有贡献,3a1轨道上的电荷发生转移,电荷转移到其他区域,包括N和Ir原子之间的电荷积累,这样也就形成了N-Ir键.对优势吸附结构的态密度(DOS)做了进一步计算,3a1轨道同金属Ir表面态之间相互作用实际上是N原子的2pz轨道同金属Ir在Z方向上的混合,也就是NH3的吸附主要是N原子的2Pz轨道与底物金属原子Ir的5dz2轨道混合作用.
猜你喜欢
中学生数理化·中考版(2021年10期)2021-11-22
保鲜与加工(2021年1期)2021-02-06
云南化工(2020年11期)2021-01-14
中学生数理化(高中版.高考理化)(2020年10期)2020-10-27
劳动保护(2018年5期)2018-06-05
数学大王·低年级(2017年9期)2017-09-18
浙江大学学报(工学版)(2016年11期)2016-06-05
新高考·高一物理(2015年6期)2015-09-28
新高考·高一物理(2015年6期)2015-09-28
小说月刊(2015年11期)2015-04-23
