
一锅法制备具有AIE效应含对苯二乙腈单元聚合物
2020-01-15 01:05梁立勋王颖高路谣陈卫平赵洪池
河北大学学报(自然科学版) 2020年1期
梁立勋,王颖,高路谣,陈卫平,赵洪池
(河北大学 化学与环境科学学院,河北 保定 071002)
近年来,唐本忠等[1-2]发现聚集诱导发光(AIE)效应,具有此效应的分子在稀溶液中,单键可以自由的转动和振动,以非辐射的形式消耗了分子激发态能量,导致荧光减弱甚至不发光,而在浓溶液或固态时,分子内旋转受到空间位阻的限制而减弱,能量衰减主要以发光的形式进行,从而使荧光增强. 将此类分子引入到水溶性聚糖中,当糖类分子与蛋白质和凝集素作用时,分子的空间位阻变大,出现荧光增强的现象,即含有AIE分子的糖基水溶性聚合物在与蛋白质特异性结合后出现荧光开启现象,这种现象可促进在活细胞内对蛋白质特异性诊断荧光探针的进一步研发[3-4].
糖类具有可再生的潜力,是生命信息承载的基础化学单元,同时也是人体能量的来源,作为生命的重要组成部分,其在生物识别过程中具有重要的作用,近年来引起了高分子化学领域研究者的广泛关注[5-8]. 含糖聚合物大多采用自由基聚合法制备,而可逆加成-断裂链转移(RAFT)聚合法作为一种活性可控聚合的方法[9-12],聚合中加入链转移试剂,使聚合反应在活性增长自由基与休眠种之间保持快速的动力学平衡,减少了自由基双基终止的机率,可调控聚合物分子量且分子量分布窄. 点击化学(Click chemistry)[13-15]具有良好的选择性、反应条件温和、反应速率快及产率高等优点.根据RAFT聚合和点击化学的特点,本文合成了α,α’-双-[对-(2,3-环氧丙氧基)亚苄基]-对苯二乙腈(M2),通过RAFT聚合制备了含双硫酯键的聚合物P1,再利用“一锅法”将P1的双硫酯键还原得到的巯基与M2的环氧基发生点击化学反应,合成含对苯二乙腈单元荧光聚合物P2,最后脱除乙酰基保护得到水溶性荧光聚合物P3.P3具有规整的结构、AIE特性、良好的水溶性和热稳定性.
1 实验部分
1.1 主要仪器和试剂
1.1.1 仪器
AVANCE Ⅲ 600 MHz超导核磁共振波谱仪(Bruker,德国);Varian 640-IR型傅里叶变换红外光谱仪(Varian,美国);Specord 210 plus型紫外-可见分光光度计(Analytikjena,德国);F-7000型荧光分光光度计(Hitachi,日本);LC-20AT型凝胶渗透色谱仪(Shimadzu,日本);Pyris 1型热重分析仪(PerkinElmer,美国);DSC8000型差式扫描量热仪(PerkinElmer,美国);Q Exactive型液质联用仪(Thermo,美国).
1.1.2 试剂
对羟基苯甲醛,醋酸酐,均为分析纯,天津科密欧化学试剂有限公司;对苯二乙腈,三氟化硼乙醚溶液,质量分数98%,甲基丙烯酸羟乙酯,甲醇钠/甲醇,质量分数30%,阿拉丁试剂公司;环氧氯丙烷,分析纯,天津市大茂化学试剂厂;硼氢化钠,天津傲然精细化工研究所;2-氰基-2-丙基苯并二硫,质量分数97%,江苏欣诺科催化剂有限公司;氘代氯仿(摩尔分数99.8%)-体积分数0.03%TMS,氘代二甲基亚砜(摩尔分数99.9%)-体积分数0.03%TMS,重水(摩尔分数99.9%),D-甘露糖,百灵威(北京)科技有限公司;硅胶(40~90 μm,试剂级),烟台银龙硅胶有限公司.
1.2 合成原理
本文合成路线如示意图1所示. 以对苯二乙腈和对羟基苯甲醛为起始原料合成α,α’-双-(对羟基亚苄基)-对苯二乙腈(M1),进而与环氧氯丙烷反应合成带有双环氧官能团的化合物α,α’-双-[对-(2,3-环氧丙氧基)亚苄基]-对苯二乙腈(M2). 醋酸酐与D-甘露糖酯化反应得到1,2,3,4,6-五-O-乙酰基-α-D-吡喃甘露糖(M3),再和甲基丙烯酸羟乙酯(HEMA)反应得到2-(2',3',4',6'-四-O-乙酰基-α-D-吡喃甘露糖基氧基)甲基丙烯酸乙酯(M4). 以2-氰基-2-丙基苯并二硫为RAFT试剂,引发M4聚合合成线性聚合物P1,再通过“一锅法”将P1的二硫酯片段还原成巯基与M2的环氧基团进行点击化学反应合成聚合物P2,最后在甲醇钠/甲醇溶液中脱除乙酰基保护得到水溶性聚合物P3.

图1 化合物和聚合物的合成路线
1.3 制备方法
1.3.1 化合物M1的合成
化合物M1参照文献[15]合成. 产物为黄绿色粉末,产率80%.1H NMR (600 MHz, DMSO-d6)δ: 10.31 (s, 2H), 7.99 (s, 2H), 7.89 (d,J=8.4 Hz, 4H), 7.82 (s, 4H), 6.93 (d,J=8.4 Hz 4H).13C NMR (150 MHz, DMSO-d6)δ:160.31, 143.07, 134.37, 131.55, 128.80, 125.94, 118.36, 115.91, 104.98. LC-MS(ESI)m/z: [M-H]-分子式是C24H15N2O2;理论分子质量363.11;实测值363.11.
1.3.2 化合物M2的合成
取M11 g (2.75 mmol)于100 mL双口圆底烧瓶中,加入5 mL环氧氯丙烷,50 mL异丙醇,70 ℃搅拌条件下缓慢滴加3.4 mL质量分数30%氢氧化钠溶液(约30 min),滴加完毕继续反应5 h,反应结束用氯仿和去离子水萃取,收集有机相并用无水硫酸钠干燥,抽滤除去无水硫酸钠,旋转蒸发除去溶剂,残留有机物用柱层析法提纯,以二氯甲烷为淋洗剂,收集第一个荧光点,旋蒸除去溶剂,真空干燥12 h得到黄绿色固体0.47 g,产率36%.1H NMR (600 MHz, CDCl3)δ: 7.91 (d,J=8.4 Hz, 2H), 7.65 (d,J=8.4 Hz, 2H), 7.51 (s, 1H), 7.47 (d,J=8.4 Hz, 2H), 7.32 (s, 1H), 7.15 (d,J=8.4 Hz, 2H), 7.02 (d,J=9.0 Hz, 2H) 6.80 (d,J=9.0 Hz, 2H), 4.34~4.24 (m, 2H), 4.03~3.91 (m, 2H), 2.93~2.43 (m, 2H), 2.95~2.90 (m, 2H), 2.80~2.73 (m, 2H).13C NMR(150 MHz, CDCl3)δ:158.91, 143.25, 141.38, 132.47, 130.65, 130.41, 128.59, 125.41, 125.36, 114.10, 113.78, 67.91, 48.92, 43.61. LC-MS(ESI)m/z: [M+Na]+分子式是C30H24N2O4Na;理论分子质量499.16;实测值499.16.
1.3.3 化合物M3和M4的合成
化合物M3参照文献[17]合成. 得到浅黄色油状液体,产率90%. 化合物M4参照文献[18]合成. 白色粉末状固体5.02 g,产率42%.M4的NMR和MS分析:1H NMR (600 MHz, CDCl3)δ: 6.11 (s, 1H), 5.58 (s, 1H), 5.34~5.32 (m, 1H), 5.26 (t,J=10.2 Hz, 2H), 4.86 (s, 1H), 4.32 (s, 2H), 4.26 (dd,J=5.4 Hz,J=12.6 Hz, 1H), 4.08~4.06 (m, 1H), 4.02~3.99 (m, 1H), 3.92~3.89 (m, 1H), 3.77~3.74 (m, 1H), 2.14 (s, 3H), 2.08 (s, 3H), 2.02 (s, 3H), 1.97 (s, 3H), 1.94 (s, 3H).13C NMR(150 MHz, CDCl3)δ: 170.58, 169.97, 119.81, 169.66, 167.08, 136.03, 126.01, 97.54, 69.44, 68.95, 68.63, 66.14, 65.90, 63.12, 62.42, 20.83, 20.67, 20.64, 18.25. LC-MS(ESI)m/z: [M+Na]+理论分子式是C20H28O12Na;理论分子质量为483.15;实测值483.15.
1.3.4 聚合物P1的合成
准确称取化合物M4937 mg (2.04 mmol)、2-氰基-2-丙基苯并二硫15 mg (0.07 mmol)和精制AIBN 3.34 mg (0.02 mmol)置于10 mL茄型瓶中,加入3.5 mL精制THF,在Ar保护下,70 ℃反应10 h,反应结束,冷却至室温,用乙醚多次沉淀聚合物,干燥得到粉红色粉末657 mg,单体转化率69%.
1.3.5 聚合物P2的合成
准确称取聚合物P1500 mg、M21.4 mg(0.003 mmol)、NaBH47 mg(0.18 mmol)和二甲基苯基膦6.34 mg(0.06 mmol)加入到25 mL茄型瓶中,加入8 mL干燥氯仿和少量无水甲醇,体系抽真空-充高纯氩气循环除氧后,磁力搅拌下室温反应96 h,反应停止后,用乙醚沉淀数次,离心后沉淀经真空干燥得白色粉末400 mg,收率80%.
1.3.6 聚合物P3的合成
准确称取聚合物P2300 mg和4 mL无水甲醇加入到25 mL圆底烧瓶中,剧烈搅拌使其溶解,随后滴加0.5 mL质量分数为30%的甲醇钠-甲醇溶液,搅拌30 min,抽滤得到白色固体,用去离子水溶解固体,抽滤除去不溶物,滤液用透析袋(MWCO:Nominal:14000)在去离子水中透析96 h. 透析液经冷冻干燥得到白色蓬松固体150 mg,收率79%.
2 结果与讨论
2.1 P2和P3的1H NMR谱图
图2为P2和P3的1H NMR谱图. 由图2可知,P3的1H NMR谱图中少了2.08、2.04、1.98和1.91处的4个乙酰基(-COCH3)上H的分裂峰,说明P2已经脱除了乙酰基,P2为油溶性聚合物而P3为水溶性聚合物,但α,α’-二亚苄基-对苯二乙腈为油溶性的,并且含量较少,所以在D2O中没有检测到苯环上H的分裂峰.
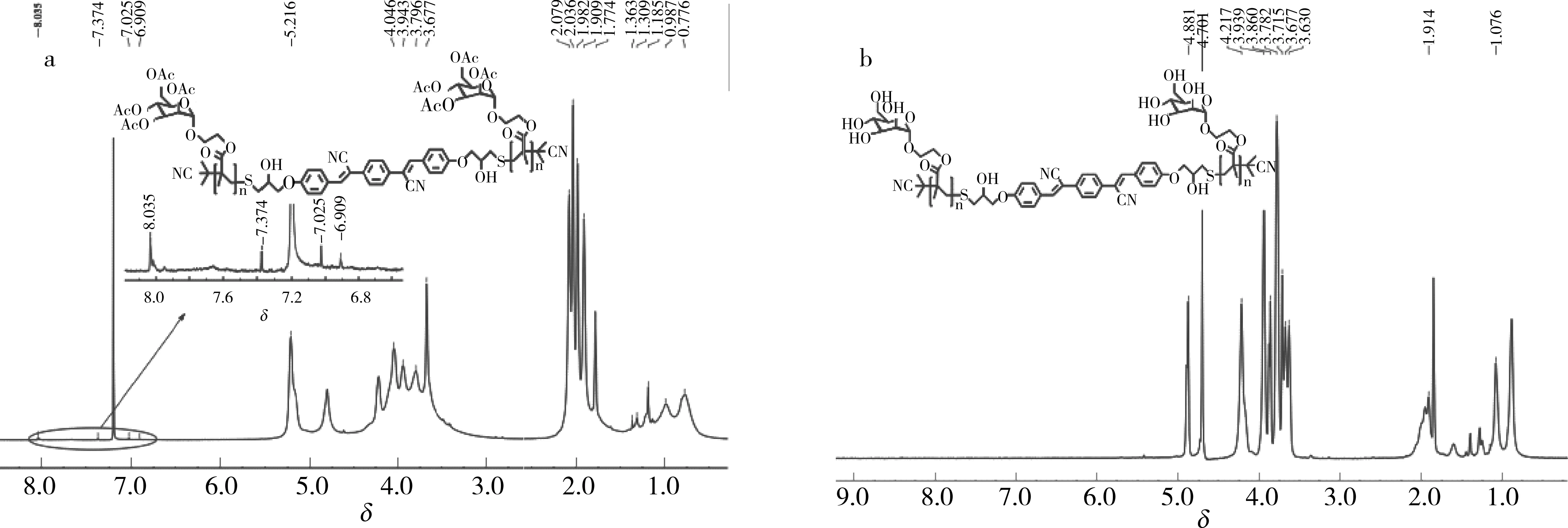
图2 (a)P2和(b)P3的1H NMR谱
2.2 FTIR分析

图3 M2、M4、P1、P2和P3红外光谱
图3为M2、M3、P1、P2和P3的红外光谱图. 由图3可知,在M2的谱图中,2 956 cm-1处是甲基C—H键的振动吸收峰,1 596 cm-1和1 513 cm-1处为苯环的骨架C—C键伸缩振动吸收峰,1 182 cm-1和911 cm-1为环氧基C—O—C键伸缩振动吸收峰. 在M4的谱图中,2 962 cm-1处是甲基C—H键的振动吸收峰,1 750 cm-1和1 234 cm-1处为羰基O=C—O键振动吸收峰,1 635 cm-1处为C=C键振动吸收峰,1 454 cm-1处为亚甲基-CH2-上C—H键的弯曲振动吸收峰. 在P1的谱图中,1 635 cm-1处的双键C=C键振动吸收峰消失,说明成功合成了聚合物P1. 在P2的谱图中,3 428 cm-1处为环氧基团开环反应后生成的O—H键振动吸收峰,并且在911 cm-1处环氧基的C—O—C键振动吸收峰消失了,说明合成了聚合物P2. 在P3的谱图中,2 931 cm-1处为甲基上C—H键的振动吸收峰,3 421 cm-1处的吸收峰显著增强,该吸收峰为聚合物P2脱去乙酰基保护后裸露出的羟基O—H键振动吸收峰,在1 750 cm-1处O=C—O键振动吸收峰减弱,说明脱除乙酰基保护合成了聚合物P3.
2.3 GPC分析
图4为聚合物P1、P2和聚苯乙烯标准样品的GPC谱图. 由图4可知,P1的保留时间比P2的长,说明P2的分子量比P1的大. 通过由聚苯乙烯标准样品确立的标准曲线计算得出:P1的数均分子量Mn=12 200 g/mol,多分散性指数PDI=1.21.P2的Mn=24 700 g/mol,PDI=2.09.
2.4 UV-Vis分析
图5为M1、M2和P3在DMSO中的紫外-可见光谱图,由图5可知,M1的紫外-可见吸收峰为379 nm,此峰对应于苯环共轭骨架上的π-π*电子跃迁.M2中引入的环氧丙氧基为供电子基团,使M2的吸收峰发生蓝移,出现在372 nm.P3存在一个从304 nm到436 nm宽而弱的吸收峰,这是因为聚合物只有中心的α,α’-二亚苄基-对苯二乙腈存在紫外吸收,但是其相对含量较少,并且聚合物主链为供电性的柔性连,使P3紫外吸收峰发生蓝移,峰值约为368 nm.

图4 P1和P2的GPC谱图

ρ(M1)=2 μg/mL, ρ(M2)=2 μg/mL, ρ(P3)= 600 μg/mL
2.5 荧光谱图分析
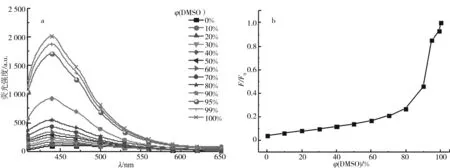
F:P3在不同DMSO体积分数时440 nm处的荧光强度;F0:P3在DMSO中440 nm处的荧光强度ρ(P3)=600 μg/mL, λex=380 nm; 狭缝宽度: 20, 20 nm
图6为聚合物P3在DMSO-H2O中荧光谱图和荧光强度比率图. 聚合物中α,α’-二亚苄基-对苯二乙腈单元是共轭分子,在苯环的转动和振动受阻时,分子内激发态能量主要以辐射方式损耗,发射出荧光,荧光发射峰在440 nm.P3荧光强度随着混合溶剂中DMSO体积分数的升高而增强,在体积分数小于80%时荧光微弱并且缓慢增强,在体积分数大于80%时荧光强度大幅度增强,表现出典型的AIE特性,荧光强度比率F/F0从0.27上升到1. 这是因为聚合物P3侧链上含有大量裸露的羟基,所以P3在水中具有良好的溶解性,聚合物链呈现舒展状态,α,α’-二亚苄基-对苯二乙腈单元中的苯环可以自由转动,分子内激发态能量主要以非辐射方式损耗,发射微弱的荧光. 而DMSO是其不良溶剂,随着混合溶剂中DMSO含量的升高,聚合物P3在混合溶剂中的溶解性逐渐降低,其聚糖分子链逐渐蜷缩,这使得分子链的空间位阻变大,α,α’-二亚苄基-对苯二乙腈单元的运动受阻,聚合物P3发生聚集,主要以发光的形式进行能量衰减,导致其荧光强度逐渐增强.
以硫酸奎宁的0.1 mol/L硫酸溶液为基准测定P3在DMSO中的荧光量子产率[20].
根据公式计算待测物的荧光量子产率,式中:Φu、Fu和Au分别表示待测物质的荧光量子产率、积分荧光强度和吸光度;Φs、Fs和As分别表示参比物质的荧光量子产率、积分荧光强度和吸光度;nu和ns分别表示待测物质所用溶剂和参比物质所用溶剂的折射率. 经计算得到P3在DMSO溶剂中的荧光量子产率为11.88%.
2.6 TGA和DSC分析
图7a是聚合物P1、P2和P3的TGA曲线(气氛:高纯氮气;气体流速:20 mL/min;起止温度:30 ℃~830 ℃;升温速率:10 ℃/min);图7b是聚合物P3的DSC曲线(气氛:高纯氮气;气体流速:20 mL/min;起止范围:-30 ℃~300 ℃;升温速率:10 ℃/min). 由图7a可知,P1、P2和P3失重率5%,热分解温度分别为317、285和323 ℃;失重率50%,热分解温度分别为391、383和378 ℃;完全热分解温度分别为455、500和486 ℃;灰分分别为3.2%、17.2%和9.7%.P1、P2和P3均具有良好的热稳定性. 由图7b可知,P3的相转变温度为118 ℃,聚合物主链为柔性链,而侧链为甘露糖类衍生物,相对于主链体积较大,减小了链的柔顺性,并且糖环上含有大量裸露的羟基形成分子间氢键,从而使相转变温度升高.
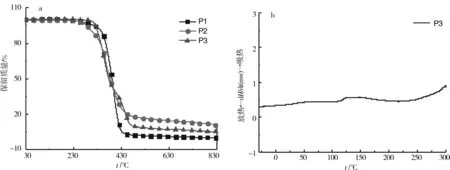
图7 聚合物P1、P2和P3的TG曲线(a)和聚合物P3的DSC曲线 (b)
3 结论
含对苯二乙腈单元水溶性荧光聚合物P3紫外吸收峰和荧光发射峰分别出现在368 nm和440 nm;其荧光强度随DMSO/H2O混合溶剂中DMSO含量升高而增强,具有典型的AIE特性;在DMSO中的荧光量子产率为11.88%.P3具有较好的热稳定性,相转变温度为118 ℃.
猜你喜欢
大电机技术(2022年5期)2022-11-17
无机化学学报(2022年9期)2022-09-16
煤化工(2022年3期)2022-07-08
农业工程学报(2022年5期)2022-06-22
化工时刊(2022年1期)2022-05-25
首都食品与医药(2020年1期)2020-10-21
世界农药(2019年4期)2019-12-30
筑路机械与施工机械化(2017年6期)2017-07-10
山东工业技术(2016年10期)2016-09-06
航天制造技术(2016年6期)2016-05-09
