
高致病性猪繁殖与呼吸综合征病毒株JXA1反向遗传平台的构建
2020-01-18 03:40韩晓亮冯松林孙彦阔王衡张桂红
华南农业大学学报 2020年1期
韩晓亮,冯松林,孙彦阔,王衡,张桂红
(1华南农业大学兽医学院,广东广州510642;2广东省动物源性人兽共患病预防与控制重点实验室,广东广州510642;3广东省兽医临床重大疾病综合防控重点实验室,广东广州510642)
猪繁殖与呼吸综合征(Porcine reproductive and respiratory syndrome,PRRS)是一种以母猪繁殖障碍和各种生长阶段猪的呼吸系统疾病为特征的病毒性传染病,俗称“猪蓝耳病”,其病原为猪繁殖与呼吸综合征病毒(Porcine rep roductive and respiratory syndrome virus,PRRSV)[1-2]。PRRS于20世纪80年代末首先在美国被发现,临床上主要表现为母猪晚期繁殖障碍和新生仔猪的呼吸道疾病[3]。随后该病相继在北美、欧洲、亚洲的养猪国家广泛流行,并引发了空前的“流产风暴”[4],给世界养猪业造成了巨大的经济损失。
2006年,在我国南方部分省份出现的“猪无名高热”病,经鉴定其病原为以JXA 1株为代表的高致病性PRRSV,引发了严重的PRRS疫情[5-6]。在PRRSV 的遗传进化树中,HP-PRRSV 和之前流行的经典毒株同属于Lineage 8,其重要特征是Nsp2编码区存在30个不连续氨基酸的缺失[7-8],这种新出现的毒株具有毒力强、致死率高、传播速度快等特点,至今依然是国内流行的优势毒株[9],给我国养猪业造成非常严重的损失。
反向遗传学技术是研究RNA 病毒的有力工具,该技术通过对相关病毒基因进行直接改造后构建出病毒的感染性克隆,并经RNA-launched 和DNA-launched 2种途径对病毒进行拯救[10-11]。RNA-launched 途径是RNA 病毒基因组先反转录成cDNA,接着克隆到合适的转录载体上,通过克隆到T7或者SP6启动子的下游,先在体外转录出病毒全长的基因组RNA,随后将这些体外的转录本转染宿主细胞,在宿主细胞内包装出活的病毒粒子[12-13]。DNA-launched 途径则是将经反转录的病毒cDNA 克隆到真核启动子如CMV 启动子的下游,并将构建好的质粒不经体外转录,直接转染宿主细胞,利用宿主细胞的核功能转录出病毒基因组,进而完成病毒粒子的包装[14]。相比RNAlaunched 途径,DNA-launched 途径避开了体外转录步骤,不仅操作简便,还降低了转染过程中RNA 降解的风险,使转染的效率大大提高。目前,反向遗传技术在研究PRRSV 的结构与功能、致病机制以及研发新型疫苗等方面有着广泛应用[15-17]。
本研究运用反向遗传操作技术将HP-PRRSV JXA 1株的全基因组分段克隆至改造过的低拷贝载体pOKq 上,成功构建了JXA 1株的全长感染性克隆。并采取基于DNA-launched 途径成功获得拯救的病毒,并对拯救病毒进行生物学特性的分析。HPPRRSV 代表株JXA 1株反向遗传平台的构建,为HP-PRRSV 后续的结构功能探索、致病机理研究以及新型疫苗研发奠定了基础。
1 材料与方法
1.1 材料
1.1.1 载体、毒株、细胞、抗体克隆载体p JET1.2购买自Therm o Fisher Scientific。由pOK 12改造具有多克隆位点的低拷贝载体pOKq、XH-GD全长感染性克隆质粒、PRRSV 易感细胞Marc-145细胞、转染用BHK-21细胞均由华南农业大学兽医学院传染病教研室保存。HP-PRRSV JXA1毒株由河南农业大学田克恭教授惠赠。PRRSV N蛋白单克隆抗体购买自韩国金诺公司。FITC标记的山羊抗小鼠IgG 二抗购买自北京中杉金桥生物技术有限公司。
1.1.2 主要试剂RNA 抽提试剂盒购自上海飞捷生物技术有限公司,反转录试剂盒、Taq酶、高保真DNA 聚合酶购自宝日医生物技术(北京)有限公司。限制性核酸内切酶购自NEB公司。T4DNA 连接酶购自Thermo Fisher Scientific。DL2000 DNA marker、250 bp DNA ladder (Dye plus)、克隆菌感受态细胞EscherichiacoliDH5α、克隆载体pMD18-T vecto r 购自宝日医生物技术(北京)有限公司;D N A 凝胶回收试剂盒、质粒抽提试剂盒购自OMEGA 公司。DMEM高糖培养基、胰蛋白酶购自Biological Industries。胎牛血清购自Gibco公司。二甲基亚砜(DMSO)购自Am resco公司。
1.2 方法
1.2.1 全长cDNA 构建方案与引物设计构建方案:JXA 1毒株全长cDNA 的构建见图1。先将CMV、A 1、A 2片段融合,融合产物再与A 3片段连接,构成A 片段。将D1片段和终止信号肽序列BGH片段融合构成D片段,然后将D、C、B、A 共4个片段依次亚克隆到低拷贝载体pOKq 上。
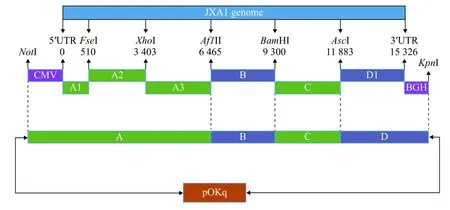
图1 JXA1毒株全长cDNA 克隆的构建策略Fig.1 The strategy for construction of the full-length cDNA clone of JXA1 strain
根据GenBank 中收录的PRRSV JXA1毒株全基因组序列,利用Oligo7.0软件设计6对引物对病毒的全长基因进行分段克隆,获得A 1、A 2、A 3、B、C和D1段。同时以PRRSV XH-GD全长感染性克隆质粒为模板,设计引物扩增CMV 和BGH 片段。同时在扩增A 1片段时引入1个沉默突变,将全基因组第510位碱基由A 突变为G,引入遗传标记FseI,以便与亲代病毒相区别。引物序列由上海英潍捷基生物技术有限公司合成。各段引物的序列、扩增片断大小及引物名称见表1。
1.2.2 JXA1毒株全基因组各片段的扩增以XHGD全长感染性克隆质粒为模板,分别用引物(CM V-F、CM V-R)和(BGH-F、BGH-R)扩增CMV 启动子序列和BGH终止信号肽序列;按上海飞捷生物技术有限公司的RNA 抽提试剂盒操作说
明书提取JXA1细胞病毒的总RNA,用适量RNase free 水溶解,获得总RNA,用SuperScriptTMIII 逆转录酶和OligodT(20)(Invitrogen,USA),按照操作说明书进行cDNA 合成,然后将获得的cDNA 作为模板,用表1中扩增引物进行各片段的扩增,经过RTPCR,获得A1、A2、A3、B、C 和D1片段,参照DNA purification kit 说明书纯化扩增的片段,利用融合PCR 技术,用引物(A 1-F、A2-R)融合A1和A 2片段,构建点突变以引入全长感染性克隆的遗传标记FseI酶切位点,以回收的CMV 片段和A1、A2融合片段为模板,用引物(CMV-F、A 2-R)融合CM V、A 1、A 2片段并回收;以回收的BGH 片段和D1片段为模板,用引物(D 1-F、BGH-R)融合D 1和BGH片段构成D片段并回收,PCR 按常规方法进行,并设立空白对照。PCR 产物用10 g/L琼脂糖凝胶(含0.5μg/mL EB)电泳检测并回收纯化。另外,为了提高目的片段的保真性,构建感染性克隆过程中的所有PCR 反应均使用宝日医生物技术(北京)有限公司的PrimeSTAR®Max 高保真DNA 聚合酶。

表1 构建JXA1株全长感染性克隆设计的引物序列Table 1 Primer sequencesused for construction of the full-length infectious clone of JXA1 strain
1.2.3 质粒pOK-A、pJET-B、pJET-C 和pJET-D的构建将CMV、A 1、A 2的融合回收产物连接到pOKq 空载体上形成载体pOK-CMV-A 1-A 2,再将扩增的A 3片段连接到pOK-CMV-A 1-A 2载体上,构建出pOK-A 中间载体。将D、C和B这3个片段的纯化产物分别与pJET1.2载体连接。取5 μL的连接产物转化克隆菌感受态细胞E.coliDH5α 后,用灭菌枪头挑取可疑菌落于含氨苄青霉素的LB液体培养基中,于37℃条件下振荡培养4~6 h,取适量菌液进行PCR 鉴定。将PCR 鉴定为阳性的菌液送上海英潍捷基生物技术有限公司进行测序验证。测序正确的重组质粒分别命名为p JET-B、p JETC和pJET-D,并按照OMEGA 公司的质粒提取试剂盒说明书进行质粒抽提。
1.2.4 JXA 1毒株全长cDNA 的构建 将pJET-D、pJET-C、pJET-B和pOK-A 按照图1所示的顺序依次亚克隆到pOKq 载体,构建的质粒经PCR 鉴定为阳性的重组菌液送上海英潍捷基生物技术有限公司进行测序,将测序正确的克隆质粒命名为pOKJXA 1,并参照OMEGA 公司去内毒素质粒提取试剂盒说明书进行去内毒素质粒抽提。
1.2.5 JXA1株病毒的拯救将JXA 1株病毒全长cDNA 的重组质粒pOK-JXA 1转染BHK-21细胞,转染使用Lipo fectam ine 3000(后文简称Lipo 3000)脂质体,具体操作步骤参照说明书。转染72 h后,将6孔板反复冻融3次,收集细胞液,4℃、7 000 r/m in 离心7m in,取上清接种于单层M arc-145细胞,吸附1 h 后补加含2%(φ)FBS的DMEM,置于37℃、CO2体积分数为5%的培养箱培养,观察细胞病变情况(Cytopathic effect,CPE),出现病变后冻融收毒,连续传12代。将拯救的病毒命名为rJXA1。
1.2.6 拯救病毒的鉴定为验证所看到的细胞病变是拯救病毒所致,排除亲本病毒污染的可能,利用引入的沉默突变FseI酶切位点进行鉴定。取第3代的病毒细胞培养悬液进行总RNA 的提取,再取2 μg RNA 进行反转录。以得到的cDNA 为模板,以A 1-F和A 2-R 为引物进行PCR 扩增。PCR产物用10 g/L琼脂糖凝胶电泳检测并回收纯化,将纯化后的PCR 产物送上海英潍捷基生物技术有限公司进行测序鉴定。
同时将拯救病毒的第3代rJXA 1-P3和亲本病毒的第3代JXA 1-P3接种至长满单层的M arc-145细胞6孔板中并设1孔阴性对照,置于37℃、CO2体积分数为5%培养箱中培养72 h 后对拯救的病毒进行间接免疫荧光(Indirect imm unofluoresence assay,IFA)试验鉴定。
1.2.7 拯救病毒的稳定性将拯救病毒rJXA 1在M arc-145细胞上连续传至12代,观察每一代的CPE情况。同时从拯救获得的病毒中,选择第1、3、6、9和12代病毒进行TCID50测定。将M arc-145细胞接种到96孔细胞培养板上,待细胞汇合度达到90%以上时,将获得的相应代次的拯救病毒进行10倍梯度倍比稀释,即用DM EM进行10−1~10−8倍比稀释,分别接种于Marc-145细胞中,每个稀释度接种8孔,每孔100μL。同时设未接毒的空白对照,将细胞板放置37℃、CO2体积分数为5%细胞培养箱作用90 min。弃去病毒液,加入含2%(φ)FBS的DMEM,放置于37℃、CO2体积分数为5%细胞培养箱。连续观察2~7 d,记录每个稀释度细胞病变的孔数,本试验重复3次求其平均值,并按照Reed-Muench 法计算各个样品的TCID50。
1.2.8 拯救病毒的生长曲线测定rJXA 1第3代病毒和亲本病毒JXA1第3代病毒在Marc-145细胞中的生长曲线。将rJXA 1第3代病毒和亲本病毒JXA 1第3代病毒用DMEM培养基稀释至感染复数(Multiplicity of infection,MOI)为0.1,接种于长成单层的M arc-145细胞,置于37℃、CO2体积分数为5%培养箱中孵育1 h,每个病毒各做3次重复。孵育结束后弃去上清,用PBS缓冲液洗涤细胞2次,加入5mL含2%(φ)FBS的DMEM细胞维持液,置于37℃、CO2体积分数为5%的培养箱中培养。分别于接种病毒12、24、36、48、60、72、84 和96 h后,从每瓶细胞中吸取200μL上清,然后测定各时相上清中病毒的TCID50,绘制病毒的生长曲线。
2 结果与分析
2.1 JXA1毒株全基因组各片段的扩增
以XH-GD全长感染性克隆质粒为模板扩增CMV 启动子序列和BGH 终止信号肽序列,以JXA1反转录后的cDNA 为模板扩增JAX 1的A 1、A 2、A 3、B、C 和D1段。PCR 产物经琼脂糖凝胶电泳后,可见相应的目的条带,其中CMV、BGH 和A 1片段的扩增产物大小分别约为750、500和500 bp,与预期大小相符(图2A)。A2、A3、B、C和D1片段的扩增产物大小分别约为2 200、3 000、3 000、2 700和3 300 bp,与预期大小相符(图2B)。
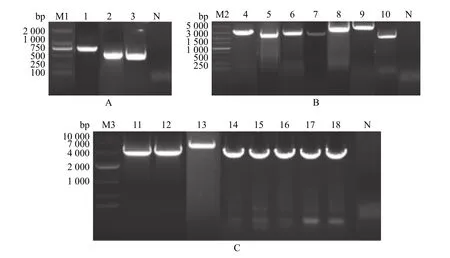
图2 JXA1株各基因片段的PCR 扩增与构建Fig.2 PCR amplification and construction of each gene fragment of JXA1 strain
利用融合PCR 技术融合A1和A2片段,构建点突变来引入全长感染性克隆的遗传标记FseI 酶切位点,以回收的CMV 片段和A 1、A 2融合片段为模板,对CMV、A1、A2片段进行融合PCR 扩增并回收,PCR 产物经琼脂糖凝胶电泳后,可见约为3 800 bp扩增片段,与预期大小相符(图2C)。将CMV、A1、A2的融合回收产物连接到pOKq 空载体上形成载体pOK-CMV-A 1-A 2,再将扩增的A3片段连接到pOK-CMV-A 1-A 2载体上,并进行菌液PCR 鉴定(图2C),构建出pOK-A 中间载体;以回收的BGH 和D1段为模板,对D1和BGH 片段进行融合PCR 扩增并回收,PCR 产物经琼脂糖凝胶电泳后,可见约为3 800 bp扩增片段,与预期大小相符(图2B),然后将D、C、B片段依次克隆到pJET1.2载体上。
2.2 JXA1毒株全长cDNA 的构建与鉴定
将pJET-D、pJET-C、pJET-B和pOK-A 按照图1所示的顺序依次连接到pOKq 载体,构建全长cDNA的重组质粒pOK-JXA1,用引物CMV-F、A1-R 对重组质粒pOK-JXA 1进行PCR 鉴定,PCR 产物经琼脂糖凝胶电泳后,可见约为1 300 bp的扩增条带,与预期大小相符(图3),鉴定阳性的重组菌液送往上海英潍捷基生物技术有限公司进行测序验证,测序正确的重组质粒命名为pOK-JXA1。
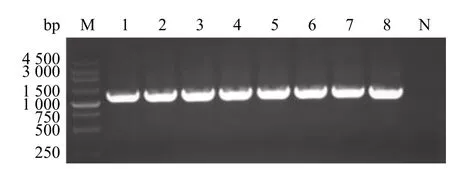
图3 pOK-JXA1菌液PCR 鉴定Fig.3 PCR identification of pOK-JXA1 bacterial liquid
2.3 JXA1株病毒的拯救
将质粒pOK-JXA1转染BHK-21细胞72 h 后,冻融收获病毒,离心取上清后接种至Marc-145细胞,置于37℃、CO2体积分数为5%的培养箱中培养72 h,显微镜下可见与亲本病毒JXA1 类似的CPE(图4),将拯救病毒命名为rJXA1。

图4 拯救病毒产生的细胞病变Fig.4 Cytopathic effect (CPE)produced in the rescued viruses
2.4 拯救病毒的鉴定
将拯救获得的rJXA1在Marc-145细胞中连续传代。取第3代病毒(rJXA 1-P3)的培养液抽提RNA 进行RT-PCR 鉴定,PCR 产物经琼脂糖凝胶电泳后可检测到强阳性条带,大小与预期相符(图5)。将纯化的PCR 产物送测序,测序结果显示拯救病毒rJXA 1全基因组的第510位核苷酸由A 突变为G,对应的阅读框由AGA 变为AGG,推导的氨基酸均为精氨酸,产生了新的酶切位点FseI(GGCCG GCC),与预期结果一致,表明拯救病毒的遗传标记被成功引入。
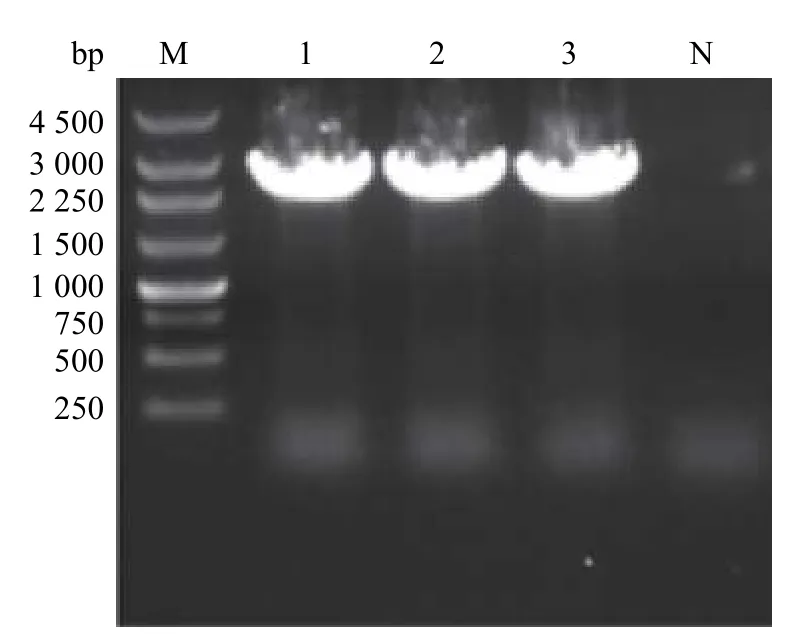
图5 拯救病毒的遗传标记位点RT-PCR 鉴定Fig.5 Identification of genetic marker of the rescued virus by RT-PCR
用PRRSV N 蛋白单克隆抗体对亲本病毒以及拯救病毒进行间接免疫荧光鉴定,结果显示:接种亲本病毒和拯救病毒的细胞在荧光显微镜下可观察到特异性的绿色荧光,而阴性对照细胞没有荧光产生(图6),证明病毒拯救成功。
2.5 拯救病毒的稳定性
将拯救病毒rJXA1在Marc-145细胞上连续传至12代,每一代均可以产生明显CPE。图7为第1、3、6、9和12代的细胞病变情况。同时对第1、3、6、9和12代病毒进行TCID50的测定,结果表明,拯救病毒可在M arc-145细胞上稳定传代,病毒滴度介于106.0~107.3TCID50/mL。
2.6 拯救病毒的生长曲线
对拯救病毒rJXA 1的第3代病毒和亲本病毒JXA 1的第3代病毒进行生长曲线的测定,结果表明拯救病毒rJXA 1第3代和亲本病毒JXA 1第3代的生长曲线相似,二者达到最高滴度的时间相同,均为72 h(图8)。

图6 拯救病毒的间接免疫荧光鉴定Fig.6 Identification of the rescued virus through indirect immunofluoresenceassay

图7 rJXA1在M arc-145细胞中引起的细胞病变Fig.7 Cytopathic effect (CPE)induced by rJXA1 in M arc-145 cells
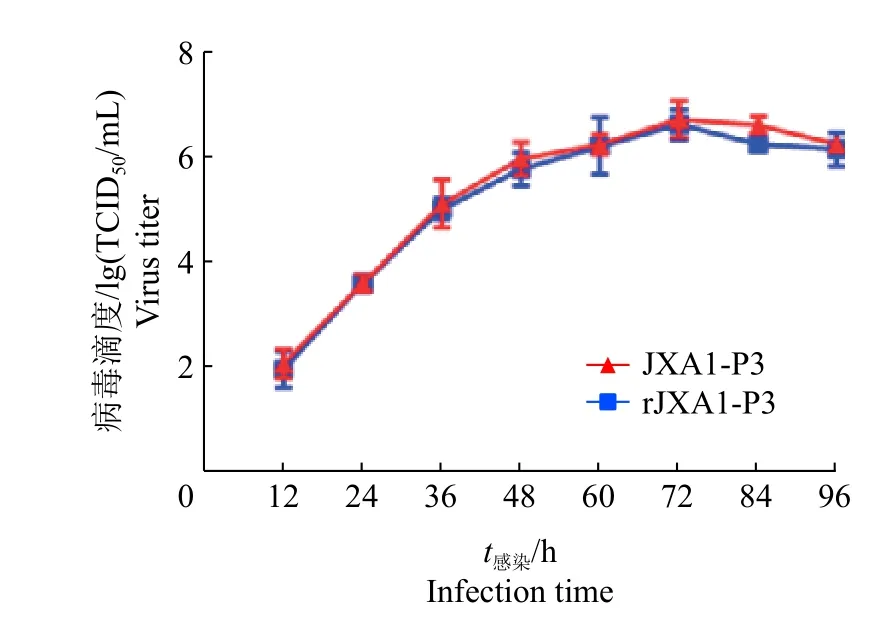
图8 拯救病毒和亲本病毒的生长曲线Fig.8 The growth curves of the rescued virus and parental virus.
3 讨论与结论
2006年以来,以感染猪高热、高发病率和高死亡率为特征的高致病性猪繁殖与呼吸综合征在我国爆发,并在全国范围流行。截至目前,该病依然在国内广泛流行,给我国养猪业带来巨大的经济损失[18]。
PRRSV 基因组为不分节段的单股正链RNA,全长15 kb左右。构建其全长感染性克隆时需要避免基因突变、缺失等破坏保真性的情况出现。为克服这一难点,本研究在构建过程中全程使用PrimeSTAR®Max 高保真聚合酶进行基因的扩增或融合PCR 扩增。此外,为了避免长片段扩增导致的保真性降低,本研究还将整个病毒的基因组分为4个节段分段克隆,逐步连接到严谨性较高的低拷贝载体pOKq 上,成功构建了JXA 1株的全长感染性克隆。并采取基于DNA-launched 途径进行病毒拯救,成功构建了HP-PRRSV JXA 1株的反向遗传平台,获得了感染性拯救病毒rJXA 1。该拯救病毒与亲本毒株JXA1具有相同的体外增殖能力。
为了区分亲本病毒和拯救病毒,需要在拯救病毒的基因中加入遗传标记。本研究选择了20株有代表性的基因II型PRRSV 毒株,使用M egA lign软件对其进行全基因组序列比对。最终选择第510位碱基作为遗传标记位点。20株基因II型代表株在该位点的碱基均为A,对应的氨基酸密码子为AGA,编码精氨酸,将其突变为G 后不影响编码的氨基酸,同时可以形成一个新的单酶切位点FseI(GGCCGGCC),便于对拯救病毒和亲本病毒的鉴定。
准确且完整的全长感染性克隆的构建是病毒拯救的关键[19]。本研究采取基于DNA-launched 途径进行病毒拯救,避开了RNA 体外转录的步骤,不仅减少了试验操作的影响,也简化了整个试验流程。另外,本研究还在JXA 1病毒的基因组两端添加了核酶序列,全长质粒转染至BHK-21细胞后在细胞内转录出RNA,在核酶的作用下直接剪切,形成不含外源基因的病毒基因组。PRRSV 基因组3′端存在1个ploy A 尾,ploy A 的个数对病毒转录的稳定性有重要作用,如果ploy A 尾上A 的个数过少,有可能导致转录失败,无法产生具有感染性的病毒粒子。基于广东省动物源性人兽共患病预防与控制重点实验室以前建立RNA-launched 反向遗传平台时的经验,本研究选择了42个A 构建病毒基因组的ploy A 尾。
虽然国内已使用高致病性PRRSV 弱毒活疫苗来控制HP-PRRSV 的流行,但HP-PRRSV 毒株依然在国内广泛流行,未呈现明显的减弱趋势[20],其他亚群(如NADC30-like 亚群和GM 2-like 亚群)呈现上升趋势[21-22],所以很有必要对其原因进行研究。本研究成功构建的HP-PRRSV JXA1株的反向遗传平台将为进一步研究HP-PRRSV 的致病机理、基因功能以及新型疫苗研发奠定了基础,同时可为PRRSV 的防控提供技术参考。致谢:感谢河南农业大学田克恭教授惠赠HP-PRRSV JXA1毒株,感谢华南农业大学兽医学院陈耀博士在本研究的选题上给予的指导与建议!
猜你喜欢
动物医学进展(2022年9期)2022-11-26
——一道江苏高考题的奥秘解读和拓展
中学生物学(2022年7期)2022-09-07
成都医学院学报(2022年4期)2022-08-19
中国典型病例大全(2022年12期)2022-05-13
昆明医科大学学报(2022年3期)2022-04-19
科学大观园(2022年2期)2022-01-23
文萃报·周二版(2021年47期)2021-12-14
心电与循环(2021年4期)2021-08-03
江西农业学报(2021年4期)2021-04-20
三农资讯半月报(2020年11期)2020-06-21
