
基于宏转录组学技术对豆酱中活菌群落分析方法的建立
2020-03-11 08:39安飞宇武俊瑞尤升波解梦汐包海燕乌日娜
食品科学 2020年4期
安飞宇,武俊瑞,尤升波,解梦汐,陈 旭,赵 越,包海燕,乌日娜,*
(1.沈阳农业大学食品学院,辽宁 沈阳 110866;2.山东省农业科学院生物技术研究中心,山东 济南 250100)
豆酱是以大豆和面粉为主要原料,经自然发酵而成的半流动状态发酵食品,在我国北方地区称为大酱[1]。传统自然发酵豆酱所具有的独特色、香、味、体是在各种微生物的共同作用下,通过诸多生化反应而形成的[2],有研究表明,细菌、霉菌和酵母菌是豆酱发酵过程中的主要微生物[3],这些微生物对于自然发酵豆酱品质的形成起到了极其重要的作用。
近年来,自然发酵豆酱中微生物群落结构在国内外得到广泛的关注和研究[4-6],同时也发展了许多研究微生物群落结构和功能的技术[7]。研究方法先后经历了传统的平板培养法[8]、基于聚合酶链式反应-变性梯度凝胶电泳(polymerase chain reaction-denaturing gradient gel electrophoresis,PCR-DGGE)技术[9-10]、构建克隆文库和DNA测序等[11-12]。近年来,随着测序技术的不断进步及测序成本的逐渐降低,科学界开始越来越多地应用高通量测序技术研究发酵食品中的微生物群落[13]。但仅在DNA层面上的测序,如16S rRNA基因测序和宏基因组测序,其结果中有很大一部分是已经死亡的微生物,测序结果不能反映不同发酵阶段菌落的真实情况,对发酵过程中实际发生的代谢反应也只能是推测[14],因此还需使用新技术对发酵体系中的活菌群落进行分析。
宏转录组学是后基因组时代最具代表性的新技术。宏转录组指的是在某个特定条件或时空,群落中所有微生物基因转录本的总和,可用于衡量微生物群落宏基因组的表达水平,筛选出高表达活性功能基因和微生物[15-16]。由于其是以微生物群落的总RNA为研究对象,因此宏转录组测序技术在一定程度上弥补了基因组测序结果存在死亡菌种干扰的这一缺陷,其结果只反映取样时体系中活菌的群落结构和相对丰度。近年来,宏转录组开始应用于如酸面团、干酪等发酵食品[17-19],用于探索发酵食品中活菌的群落结构及功能,并通过分析各活菌群落在发酵过程中的代谢途径揭示其对风味的贡献,而由于自然发酵豆酱体系复杂,其总RNA的提取存在一定困难,目前鲜见有将宏转录组学技术应用于发酵豆酱的报道。
本实验基于宏转录组学技术对自然发酵豆酱初期和末期的两个样本进行活菌群落检测,建立了适用于豆酱这一复杂体系的总RNA提取方法,并对比分析了自然发酵豆酱在不同发酵时期的活菌群落结构及功能,以期为揭示豆酱发酵过程中真正发挥作用的微生物和优良发酵菌种的选育提供基本的技术支持,也可作为其他半固态发酵食品菌群分析的技术参考。
1 材料与方法
1.1 材料与试剂
RNA PowerSoil®Total RNA Isolation Kit美国Mobio公司;Ribo-Zero Magnetic Gold Kit 美国Epicentre公司;TruSeq Stranded mRNA LT Sample Prep Kit美国Illumina公司;High Sensitivity DNA Kit美国Agilent公司。
1.2 仪器与设备
NanoDrop分析仪 美国Thermo Scientific公司;琼脂糖凝胶电泳仪 北京市六一仪器厂;Centrifuge 5804R离心机 德国Eppendorf公司;2100生物分析仪 美国Agilent公司;HiSeq测序系统 美国Illumina公司。
1.3 方法
1.3.1 样品采集
样品取自辽宁省沈阳市辽中区一制酱经验丰富的农家,分别采集发酵第0天和第56天的豆酱样品于无菌灭酶管中,后续采用两种不同的样品前处理方式:1)采样后低温运输回实验室,4 ℃、8 000 r/min离心5 min,弃上清液,重复处理一次后再液氮速冻并转移至-80 ℃冰箱保存;2)采样时直接液氮速冻并转移至-80 ℃冰箱保存。
1.3.2 RNA提取与质检
采用RNA PowerSoil®Total RNA Isolation Kit,抽提总RNA,对抽提完成的RNA样品,进行1.5%琼脂糖凝胶电泳进行质量判断,要求RNA条带完整,无降解。利用紫外分光光度计对RNA进行定量。
马思特(上海)化学有限公司是总部在美国的Master Chemical Corporation设立的金属切削液及相关设备专业制造商,于2002年在上海注册成立的。马思特(上海)化学有限公司是TRIM牌金属切削液产品在亚洲地区的生产、研发和技术服务中心,也是为机加工行业提供化学品专业管理的服务商。
1.3.3 宏转录组文库构建及测序
首先将rRNA序列特异性的探针与总RNA杂交,然后利用磁珠去除rRNA探针复合物,最后再用乙醇沉淀法进一步纯化mRNA。使用Illumina公司的TruSeq Stranded mRNA LT Sample Prep Kit试剂盒进行文库的构建。包括RNA片段化、cDNA合成、cDNA文库构建、PCR扩增已经加上接头的DNA片段、文库片段选择与纯化等步骤,各步骤遵照试剂盒生产商的操作指导完成。
用2100生物分析仪对文库进行质检,合格的文库应该有单一的峰,无接头。然后,在Promega QuantiFluor上对文库进行定量,合格的文库计算后浓度应在2 nmol/L以上。最后对合格的文库在Illumina HiSeq X-ten平台上进行2h150 bp的双端测序。将需要上机的文库梯度稀释,并按所需数据量比例混样。混好的文库变性成单链进行上机测序。
1.4 数据处理
首先对测序下机的原始数据进行筛查和过滤。质控后对每个样本的mRNA序列,分别采用Trinity进行从头组装拼接[20]。将样本序列与NCBI-NT数据库中的细菌、古菌、真菌和病毒序列进行BLASTN比对。运用MEGAN软件进行物种分类注释[21]。最后结合序列在各样本中的丰度数据,使用QIIME软件获得各样本在各个分类等级上的相对丰度分布表并绘制柱状图[22]。对各样本组装拼接后得到的所有转录本以0.95的相似度和0.9的最低覆盖度进行归并去冗余,并以最长的序列作为该Unigene的代表序列。将Unigene集与KEGG及CAZy数据库比对,从而对转录本功能进行注释分析。
2 结果与分析
2.1 总RNA琼脂糖凝胶电泳分析
由于豆酱样品中的大豆经过高温高压蒸煮并长时间发酵,因此提取物中不含植物源RNA。此外,自然发酵豆酱体系复杂且处于开放式的发酵环境,豆酱中存在大量的核糖核酸酶,其会对总RNA的提取造成影响,使得从豆酱中提取RNA存在一定难度。因此与传统动植物组织及发酵食品总RNA提取方式[23-26]不同,本实验尝试不同的前处理方式,通过对总RNA抽提效果的比较建立最适用于豆酱这一复杂体系的总RNA提取方法。琼脂糖凝胶电泳检测结果如图1所示,不同前处理方式所获得的RNA质量有明显的差异。同时,不同发酵时期的总RNA提取效果也存在一定差异。
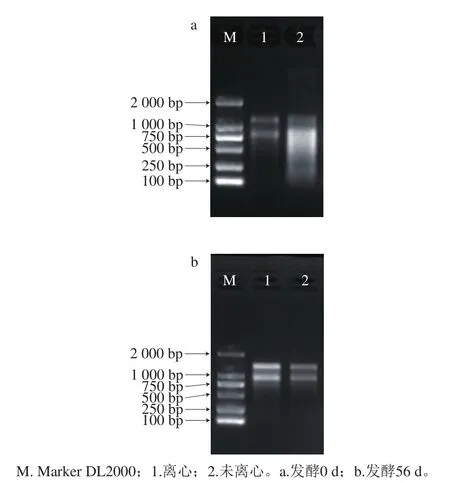
图1 两种不同前处理方式提取豆酱总RNA的电泳图Fig. 1 Electropherograms of total RNA extracted from soybean paste by two different pretreatment methods
如图1所示,经过离心处理后的样品能得到23S、16S条带,条带清晰,无明显拖尾现象,且23S的亮度高于16S条带,说明经离心处理后提取的总RNA完整性较好,纯度较高,可用于后续实验;而未经离心处理的样品RNA条带相对较暗,尤其是发酵第0天条带拖尾严重,完整性较差,无法满足宏转录组建库要求。同时实验表明,发酵第0天的豆酱电泳条带亮度明显弱于发酵第56天,说明发酵初期豆酱所抽提的RNA浓度可能低于发酵末期。
2.2 总RNA质量分析

表1 RNA检测结果Table 1 Quality evaluation of total RNA
使用2100生物分析仪检测RNA样品的纯度、质量浓度和完整性(表1)。除有明显拖尾的H0(b)外,样品通过离心处理所提取的RNA质量浓度较高,分别为30.45、44.68 ng/μL。该结果与琼脂糖凝胶电泳检测结果一致,说明发酵初期豆酱所抽提的RNA质量浓度低于发酵末期。其可能的原因为,在发酵第0天豆酱体系刚刚混入盐水,水分含量较高,并且发酵环境的巨大改变也可能会造成RNA降解。到发酵后期,豆酱水分含量降低且豆酱体系稳定,因此抽提相对容易。
纯度较好的RNA其OD260nm/OD280nm值应介于1. 8~2.0。若OD260nm/OD280nm值小于1.8,则表明多糖、蛋白和酚类物质较多,若OD260nm/OD280nm值大于2.0,则表明RNA有部分降解[23]。本实验各样品所抽提RNA的OD260nm/OD280nm值在1.90~2.11之间,其中通过离心处理的样品所抽提的RNA降解较少;而OD260nm/OD280nm值均低于1.0,说明可能仍有部分杂质残留,但不影响后续文库的建立。除未经离心处理的H0样品外,RNA完整数目(RNA integrity number,RIN)值均大于7,所提取的RNA完整性较好。检测结果表明,经离心处理提取的RNA质量浓度纯度和总量均符合转录组测序要求。
2.3 测序数据统计及序列组装
宏转录组测序涉及到扩增环节的有两个步骤,一个是RNA反转录cDNA时的逆转录-PCR,一个是测序过程中的桥式PCR,均不具明显的扩增偏好性。同时,为保证后续分析质量可靠,对测序下机的原始数据进行筛查和过滤,获得可用于后续分析的有效序列集(Clean Data),并统计其占测序原始数据的比例。其结果如表2所示。去除低质序列后,各个样品的Clean Data获得率均在96%以上,Clean Reads获得率达到99%以上,数据质量足够满足分析需要。

表2 测序数据统计Table 2 Statistical analysis of sequencing data
在得到高质序列后由Trinity软件进行拼接组装[19],相应统计结果如表3所示,不同发酵时期豆酱分别获得154 922 条和22 552 条序列,且拼接效果良好。

表3 组装结果统计Table 3 Statistics of assembly results
2.4 宏转录组物种注释和丰度分析
将样本序列与NCBINT数据库进行比对。使用Krona软件进行菌群分类学组成的交互展示[27](图2),图中圆圈从内到外依次代表界、门、纲、目、科、属6 个分类水平,扇形的大小反映了不同分类单元的相对丰度高低。在每个分类水平,各单元以不同的颜色加以区分。
如图2所示,相对丰度最高的门类为厚壁菌门(Firmicutes)。发酵初期(H0)豆酱的活菌群落结构较为复杂,除厚壁菌门(81.6%)外,Mucoromycota(3.8%)和子囊菌门(Ascomycota,3.8%)为主要菌门。在发酵末期(H56)厚壁菌门(Firmicutes)占绝对优势,其相对丰度为99.0%。在属水平上,自然发酵豆酱的主要微生物菌属为四联球菌属(Tetragenococcus)、乳杆菌属(Lactobacillus)、葡萄球菌属(Staphylococcus)、太平洋海洋杆菌属(Oceanobacillus)和肠球菌属(Enterococcus),均为细菌菌属。在发酵初期,其活菌群落结构相对复杂,除相对丰度最高的四联球菌(Tetragenococcus,34.6%)外,还鉴定出大量的葡萄球菌属(Staphylococcus,24.9%),但随着发酵的进行,葡萄球菌属相对含量迅速降低,说明其可能为制酱初期所引入的杂菌,对豆酱的发酵作用影响不大。此外,在发酵初期有一定数量的真菌被检出,如根霉菌属(Rhizopus,3.06%)、曲霉菌属(Aspergilus,2.71%),但到了发酵末期,真菌菌属丰度大幅降低,自然发酵豆酱中活菌群落结构趋于稳定,主要的菌属为四联球菌属(Tetragenococcus,70.9%)和乳杆菌属(Lactobacillus,20.0%)。四联球菌属是促进健康的益生菌,它广泛存在于豆酱、酱油、鱼酱等发酵食品中,在改善豆酱风味方面具有重要作用[28]。有研究表明[29],豆酱中的乳杆菌主要是植物乳杆菌,其不仅有利于食品的发酵,还可以改善食物的风味,且具有一定益生特性,包括维持肠道内菌群平衡、抑制肿瘤细胞的形成、降低血清胆固醇等[30]。此外,有部分序列被注释为病毒,但其相对含量极低,一些潜在致病菌如葡萄球菌属(Staphylococcus)也仅在发酵初期才有较高丰度,其含量会随着发酵时间的延长而降低,因此认为豆酱不具消费风险。本实验证明宏转录技术完全可以用于自然发酵豆酱活菌群落结构分析,而且此技术具有一定的敏感性,可以显示出豆酱发酵初期与末期间活菌群落的差异,同时也证明豆酱在自然发酵过程中微生物群落演替是一个逐渐趋于稳定的过程。

图2 基于Krona的分类学组成信息交互展示图Fig. 2 Krona-based taxonomic composition information interactive display
2.5 宏转录组功能注释和丰度分析
2.5.1 KEGG功能注释
基于各Unigene在KEGG数据库中的功能注释结果,使用QIIME软件,获取各样本对应于KEGG数据库第1和第2功能等级的相对表达量分布,其结果如图3所示。在第1等级的代谢通路分类中(图3A),代谢表达量占比最高,达到总体表达量的35.9%。发酵0 d时,代谢功能占其总表达量的27.5%,后随着发酵的进行迅速升高,发酵第56天时,其相对表达量为44.2%。

图3 KEGG数据库相对表达量分布统计图Fig. 3 KEGG database relative expression distribution statistics at (A)fi rst and (B) second levels
进一步对代谢通路子功能进行分析,如图3B所示,在第2等级的代谢通路分类中,碳水化合物代谢功能表达量占比最高,达到总体表达量的18.4%。在发酵第0、56天的相对表达量分别为12.0%和24.8%,这表明自然发酵豆酱菌群对淀粉具有较高的利用率。其中糖酵解/糖异生和TCA循环所注释到的表达量最高,分别为总体表达量的4.5%和3.7%。微生物可通过糖酵解途径将糖类分解成乙醇和二氧化碳[31],其有助于豆酱的风味产生,因此在发酵后期该功能表达量升高。此外,氨基酸代谢功能表达量也相对较高,达到总体表达量的4.7%,在发酵第0天及第56天的相对表达量分别为3.4%和6.0%。该结果与其他组学研究结果一致[32-34],说明宏转录技术可以用于分析自然发酵豆酱活菌群落功能。
2.5.2 CAZy功能比较分析
根据KEGG数据库的对比结果发现自然发酵豆酱中的微生物活动大多与碳水化合物代谢有关,因此后续进行CAZy注释分析。基于不同发酵阶段样本在CAZy功能数据库中所注释到的相对表达量,可以比较它们在两样本间的倍数差异关系,从而评估各样本所分别富集的功能类群。如图4所示,除糖苷水解酶和糖基转移酶在H0中有略高表达量外,其他4 个功能类群包括碳水化合物结合模块、辅助氧化还原酶、碳水化合物酯酶以及多糖裂解酶均在H56中有较高的活性表达。

图4 CAZy蛋白模块比较分析图Fig. 4 Comparative analysis of CAZy protein modules
微生物产生的糖基转移酶和糖苷水解酶能生成低聚糖。相关研究表明,发酵末期有机碳和可溶性糖含量都逐渐下降并保持恒定[35],说明糖苷水解酶及糖基转移酶相关代谢反应已经基本完成。此外,由于在发酵前期盐浓度较高且含氧量较低,霉菌已经基本停止生长,但由霉菌分泌的各种酶类仍继续发挥作用[36],而到了发酵末期,霉菌丰度极低,无法分泌相关酶类,这可能是发酵末期糖苷水解酶和糖基转移酶表达活性降低的主要原因。该结果也与宏转录组物种注释结果一致,再次印证宏转录技术可以用于分析自然发酵豆酱活菌群落结构及功能,且分析结果准确。
3 结 论
近年来,自然发酵豆酱中微生物群落结构及功能在国内外得到广泛的关注[4-6],为反映自然发酵豆酱菌落的真实情况,免除传统分子生物学手段所带来的死亡菌群干扰,故尝试使用宏转录组学技术对豆酱发酵体系中的活菌群落进行分析。
本实验首先对豆酱总RNA提取的前处理方法进行了对比研究,结果表明,与传统植物组织或发酵食品样品前处理方式不同[23-26],对豆酱样品进行离心处理可以很好地解决复杂发酵体系对RNA提取的影响,这种样品前处理方式对发酵初期豆酱总RNA提取的帮助尤为明显。然后对豆酱活菌菌群结构及功能进行注释,根据NCBI的注释结果,以相对丰度不低于0.01为阈值,共鉴定出13 个属的15 种微生物,其中细菌占绝大多数,主要的细菌属为四联球菌属(Tetragenococcus)和乳杆菌属(Lactobacillus)。根霉菌属(Rhizopus)、曲霉菌属(Aspergilus)仅在发酵初期有较高丰度,其可能是由酱醅引入的,但随着发酵的进行,真菌丰度大幅降低,说明在酱醪发酵阶段,细菌为优势微生物,在发酵过程中起主导作用。其物种注释结果与先前报道可以相互印证[4,6,9-10],证明宏转录技术完全可以用于自然发酵豆酱活菌群落结构分析,在显示出豆酱发酵初期与末期间活菌群落的差异的同时,也证明豆酱在自然发酵过程中微生物群落演替是一个趋于稳定的过程。在功能注释方面,通过KEGG功能注释和CAZy功能比较分析,发现自然发酵豆酱中微生物功能主要集中在碳水化合物和氨基酸代谢,其中在真菌丰度较高的发酵初期,糖苷水解酶和糖基转移酶相关代谢反应活性较高,该结果也与宏转录组物种注释结果一致,再次印证宏转录技术可以用于分析自然发酵豆酱活菌群落结构及功能,且具有良好的灵敏度及稳定性。将该技术与其他组学技术数据整合,将有利于挖掘出更多的有益菌株和潜在的功能基因应用于未来的工业生产,同时还可将该实验体系应用于其他半固态发酵食品的活菌群落分析。
猜你喜欢
中国调味品(2022年9期)2022-08-30
中国典型病例大全(2022年7期)2022-04-22
河南畜牧兽医(2021年9期)2021-12-10
餐饮世界(2021年10期)2021-11-20
猪业科学(2021年3期)2021-05-21
幽默大师(2020年10期)2020-11-10
中华诗词(2019年1期)2019-11-14
猪业科学(2018年4期)2018-05-19
中国调味品(2017年2期)2017-03-20
中国酿造(2016年12期)2016-03-01
