
基于固体核磁共振方法的蛋白质组装体三维结构解析
2020-04-24 05:42邓静马涛常自伟赵伟静杨俊
物理化学学报 2020年4期
邓静 ,马涛 ,常自伟 ,赵伟静 ,杨俊 ,*
1中国科学院武汉物理与数学研究所,波谱与原子分子物理国家重点实验室,武汉磁共振中心,武汉 430071
2中国科学院大学,北京 100049
1 引言
蛋白质组装体广泛存在于生物体内,并行使不同的生物学功能,如有些蛋白质组装体参与组成细菌分泌系统1、病毒衣壳2和细胞骨架细菌素细丝3等,而某些蛋白质组装体存在于病变的生物组织或细胞内,参与某些疾病的发生或发展,如与神经退行性疾病密切相关的淀粉样蛋白纤维PrP(Prion Protein)4、Aβ (Amyloid beta)5和α-syn (αsynuclein)6等。因此,对于这些组装体的研究有利于理解其生物学功能及相关疾病的发生发展。目前,常用的解析蛋白质高分辨三维结构的方法包括X射线晶体衍射(X-ray),核磁共振技术(包括液体NMR,固体NMR),和冷冻电镜(Cryo-EM)等。其中,固体NMR是一种强有力的分析手段,广泛应用于物理、化学和生物等领域7。在蛋白质组装体的结构解析中,与其它方法相比,固体NMR具有以下独特优势:(1)蛋白组装体分子量大,且有些样品如淀粉样纤维溶解度很低,无法使用液体NMR技术进行研究。而固体NMR适用于难溶或不溶的样品,且信号线宽不受蛋白质分子量大小影响,因此擅长于蛋白质组装体的研究;(2)蛋白质组装体难结晶,限制了X射线晶体方法的应用;而固体NMR不需要样品结晶,可在更接近生理条件下探测其结构信息;(3)近年来Cryo-EM广泛应用于蛋白质结构解析,它可快速获得蛋白分子整体轮廓,但要得到高分辨的三维结构仍有一定难度。固体NMR可获得原子水平的蛋白结构信息,常与Cryo-EM联用共同完成对蛋白质组装体的高分辨结构解析。如今固体NMR及其联用技术应用于多种蛋白质组装体的结构研究中,成功获得了一些蛋白质组装体的高分辨结构,如图1中的生物组装体结构和图2中的淀粉样纤维结构。
固体NMR可以测定蛋白质组装体的二级,三级和四级结构信息,在测定其高分辨结构时一般遵循以下流程:(1)制备蛋白质组装体样品。(2)完成蛋白质氨基酸残基的化学位移归属。基于13C检测的固体NMR多维实验(如三维NCACX,NCOCX和CONCA等)采集得到残基内和残基间主/侧链相关信号,通过主链行走的办法完成残基化学位移归属。(3)采集结构约束信息。固体NMR中常利用重耦技术(包括13C–13C同核重偶和15N–13C异核重偶)得到空间距离接近的原子间相关信号,对这些信号进行指认,得到距离约束信息。此外,固体NMR常联合其它实验技术(如STEM,X-ray和Cryo-EM等)收集重要的结构辅助信息。(4)蛋白质组装体结构计算与优化。以收集到的所有结构约束信息为依据,利用结构计算软件如XPLORNIH11、CNS12、CYANA13等对蛋白质组装体高分辨结构进行计算与优化。

图1 固体NMR解析的生物组装体的高分辨结构示意图Fig. 1 Examples of biological assemblies that have resulted in high resolution structures by solid-state NMR data.

图2 固体NMR解析的淀粉样蛋白纤维高分辨结构示意图Fig. 2 Examples of amyloid protein fibrils that have resulted in high resolution structures by solid-state NMR data.
在过去近十年中,通过固体NMR解析生物大分子结构的数量显著增加,在这些结构中,大约30%属于蛋白质组装体18,表明固体NMR在蛋白质组装体的研究中发挥着越来越重要的作用。在固体NMR对蛋白质结构的解析中,关键步骤是收集距离约束条件,距离约束条件的数量和质量决定了最终三维结构的质量。而良好的谱图分辨率是获得距离约束条件的重要因素,因此提高谱图分辨率是解析蛋白质三维结构的重要前提。本文介绍了固体NMR中改善谱图分辨率和收集蛋白质结构约束条件的常用方法,以及固体NMR与其它技术的联用解析蛋白质组装体的方法,并以Aβ淀粉样蛋白纤维和T3SS针状体结构解析为例介绍固体NMR在蛋白质组装体结构解析方面的进展。
2 利用固体NMR技术获得蛋白质结构约束信息
2.1 改善谱图质量的方法
信噪比和分辨率是评价谱图质量的两大要素。高信噪比一般通过13C/15N同位素标记和提高样品量的方法获得。谱图分辨率一般以信号线宽(即半高宽)作为评判标准。影响信号线宽的因素包括均匀增宽和非均匀增宽。粗略地讲,均匀增宽是指每个核对线宽的贡献都相同,非均匀增宽中每个核对线宽的贡献不同,影响因素包括同核偶极-偶极耦合,异核偶极-偶极耦合和化学位移各向异性等。在固体NMR中蛋白样品运动受限,不能如溶液状态一样通过自身快速翻转平均偶极-偶极耦合和化学位移各向异性,故而信号线宽增宽严重。快速魔角旋转(MAS)可有效压制偶极-偶极耦合和化学位移各向异性作用,大大提高谱图的分辨率。为了进一步提高谱图质量,在样品制备和同位素标记方法上进行了不遗余力的探索,为结构距离约束的采集奠定基础。
2.1.1 优化样品制备方法
蛋白质构象不均一会引起谱线的非均匀增宽,在固体NMR中是影响谱图分辨率的主要因素。在蛋白质纤维样品的研究中发现,改变制备样品的条件就可能得到不同构象的纤维。可通过优化纤维孵育时间,搅动速度,温度,pH,蛋白浓度,缓冲液性质和其它物质(如金属离子、磷脂膜19、螯合剂、氧化还原剂、抗菌分子等)的存在与数量等来提高样品的构象均一性。样品的水合程度也会影响谱图分辨率,因此蛋白样品装入转子时的状态(水合还是冻干,以及含水量)也需要优化。
2.1.2 优化样品同位素标记方法
同位素标记的蛋白样品可以通过化学合成或生物表达的方式获得,而对于一般蛋白样品,通过基因重组表达(大肠杆菌,酵母或其它真核细胞)是获得15N/13C标记蛋白样品的有效方法。标记方法包括均匀全标记(uniform labeling),稀疏标记(sparse labeling)和选择性标记(selective labeling)等。均匀全标记样品谱峰重叠严重,且存在很强的偶极截短效应20。偶极截短效应是指当周围标记的原子数目多时,邻近的原子的偶极耦合占主导作用,信号不能实现较远距离传递,不利于长程距离约束的获得。稀疏标记方法利用特殊13C源选择性的标记某些原子,如1-13C葡萄糖碳源表达的蛋白质倾向标记丝氨酸和丙氨酸Cβ,而2-13C葡萄糖碳源倾向标记丝氨酸和丙氨酸Cα。这种方法有效的降低了偶极截短效应,同时与均匀标记相比,信号线宽明显下降,因此可以大大提高谱图分辨率。此外选择性标记可特异性的标记某类氨基酸,甚至是某个原子,实现特定位点的信息采集。
2.1.3 其它方法
通过改善实验条件和实验方法也可以提高谱图的质量,如采用更高磁场的谱仪(高达1 GHz),不断优化的极化转移技术21,采用超快速MAS探头22以及高转速下的1H检测实验方法23对信号的灵敏度和谱图分辨率都有很大改善。我们课题组提出了新的脉冲序列,可增强脂肪族13C/13C同核间24和13C/15N异核间25极化转移效率,提高信号强度,节省固体NMR多维实验的时间。
2.2 提取结构约束的方法
蛋白质结构的质量依赖于结构约束的数目和质量。结构约束条件主要包括角度约束和距离约束,距离约束又分为单体内(或分子内)和单体间(或分子间)的距离约束。这些结构约束可确定蛋白质单体折叠方式以及单体堆叠的界面信息。本节主要介绍固体NMR获取结构约束条件的常用方法和一些新的技术手段。
2.2.1 角度约束
蛋白质的主链扭转角信息(如φ/Ψ二面角)作为结构计算中重要的角度约束,可通过TALOS+26利用氨基酸的13Cα,13Cβ,13C’和15N的化学位移预测得到。此外根据相关公式27计算其二级化学位移可推断残基片段的二级结构类型(如α-螺旋,β-折叠或无规结构)。
2.2.2 距离约束
距离约束对蛋白质结构解析至关重要,尤其是长程距离约束,涉及的实验方法有13C-13C相关的PDSD28、DARR29、PAR30等,13C-15N相关的REDOR,TEDOR31等,以及1H-1H相关的NHHC,CHHC实验32,可采集到空间距离邻近的13C/13C原子间或13C/15N间的相关信号,即为距离约束。改变混合时间可以控制信号传递的距离范围,在不考虑弛豫时,混合时间越长,信号传递的越远,越有利于建立长程耦合。但是,CHHC/NHHC、PAR/TEDOR和PDSD/DARR的混合时间通常分别小于1.0、20和500 ms。这是因为过长的混合时间既使信号因弛豫而衰减,也使信号因传至更多自旋而分散,反而不利于获取长程相关信息。相关信号一般分为四类:(1)残基内13C-13C的相关,即所有相关信号来自同一个残基;(2)短程相关,来自残基i与(i ± 1) –(i ± 2)的相关;(3)中程相关,包括从残基i与(i ± 2)–(i ± 4)的相关性。(4)长程相关/远程相关,包含残基i与> (i + 4)的相关,较多出现在长混合时间谱中(如600M谱仪中混合时间300–500 ms等)。长程相关的残基在一级序列中距离较远,但空间距离较近(约1–9 Å),这对于蛋白折叠方式的确认十分重要。
在实际谱图分析中,很多信号归属不明确,即模糊约束(ambiguous restraints),归属的不确定性主要源于两方面:谱图分辨率很差,信号重叠无法区分;单体内残基与单体间残基相关信号同时出现,难以区分。谱图的分辨率和信号重叠可以通过优化样品制备或标记方法进行改善,而对于单体内与单体间残基信号的区分,则通过设计同位素的标记方式进行区分,详细内容见章节4.1,图3b。
2.2.3 其它获得距离约束的方法
氢检测技术近年来得到快速发展,在固体NMR中的应用逐渐成熟。由于1H比13C原子具有更高的旋磁比(约是13C原子的4倍),同时1H具有几乎100%的天然丰度,且在蛋白质中含量高分布广,因此1H非常适用于蛋白长程距离约束测定。目前,高磁场核磁谱仪的应用,快速MAS技术的发展和样品氘代方法的设计都极大促进了氢检测技术在固体NMR中的发展33。氢检测技术提高了谱图分辨率和灵敏度,减少了蛋白质样品用量和实验时间,得到丰富的结构约束条件,在生物大分子的结构解析中具有强大潜力。
顺磁标记技术是基于未成对电子与自旋核之间的偶极–偶极相互作用而建立起来的技术。未成对电子自旋可以产生磁场,且电子的旋磁比高于自旋核~2–3个数量级,因此自由电子对与自旋核间具有很强的偶极-偶极耦合作用。这种相互作用的辐射范围最远可达20–24 Å,远大于传统的自旋核间相互作用的距离上限(约为9 Å)。在固体NMR中引入顺磁标记技术可获得更为丰富的距离约束条件。Sengupta等34利用顺磁驰豫增强(PRE,Paramagnetic relaxation enhancement)技术,引入Cu2+顺磁探针在固体NMR中得到丰富的距离约束信息。此外我们课题组35结合赝接触位移技术(PCS,Pseudocontact shift)和Rosetta方法,通过引入的磁各向异性顺磁金属离子获得了固体NMR的PCS数据,并计算得到GB1蛋白的高分辨三维结构。因此,利用顺磁标记技术可以为固体NMR解析蛋白结构提供新思路。
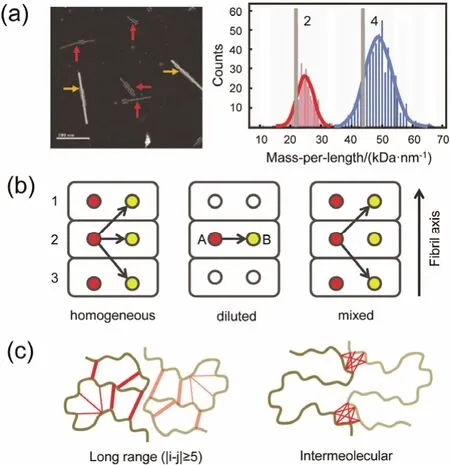
图3 Aβ42纤维结构解析相关的距离约束Fig. 3 The structure restraints determining the Aβ42 fibril structure.
3 固体NMR联合其它技术解析蛋白质结构
近年来,固体NMR与其它研究手段的联用得到快速发展。这些研究手段包括X-ray晶体衍射,STEM,Cryo-EM等实验技术以及CS-Rosetta和分子动力学(MD,molecular dynamics)模拟等结构预测和模拟手段。X-ray晶体衍射常见于晶体样品的结构解析,也应用于一些结晶淀粉样蛋白中,揭示了其交叉β折叠的结构特性,且发现其β片层间的距离约为4.7 Å,后证明该特性广泛存在于淀粉样纤维中36。故在淀粉样蛋白中,固体NMR常结合X-ray晶体衍射的结论共同描述其三维结构37。STEM可提供蛋白质组装体的MPL数据,有助于确认单体堆积方式38,详细过程见章节4.1。MD利用分子的力场约束和能量约束来演示分子运动,尤其在描述瞬态结构或结构中间态发挥着重要作用,常与固体NMR技术联用确认蛋白的结构模型以及其动力学信息39。
氨基酸的化学位移中包含了丰富的蛋白质构象信息。Rosetta等40是一种结合化学位移数据进行蛋白质结构预测的软件。Rosetta把目标蛋白质序列分为若干个片段,从蛋白质数据库(PDB)中搜索序列相似的结构,并加入实验所得的化学位移数据对所选结构进行筛选,提高所选结构的可信度。Rosetta结合PDB中的结构信息与实验数据对目标蛋白进行模拟,可快速获得与实验条件更为接近的结构模型或高分辨结构。如在细菌衣壳蛋白M138的结构确认中,基于明确的固体NMR距离约束利用Rosetta得到一个结构模型,在该结构模型基础上对模糊的距离约束进行归属,最后得到高分辨结构(RMSD为0.47 Å)。
Cryo-EM是近年来热门的蛋白结构解析手段,固体NMR与Cryo-EM联用技术逐渐广泛地应用于多种蛋白质组装体的结构解析。最具有代表性的是T3SS针状体的高分辨结构41的确认,结合Cryo-EM得到的7.7 Å的密度图和固体NMR的二面角约束和大量距离约束条件,利用Rosetta将两套数据整合得到了分辨率为0.4 Å的高分辨结构,详细过程见章节4.2。这种固体NMR-Cryo-EM的联用技术也被应用到淀粉样纤维的结构解析,如运甲状腺素蛋白的交联淀粉样蛋白原纤维中,将固体NMR测量的分子内距离约束和扭转角约束与Cryo-EM的电子密度图结合,解析得到了运甲状腺素蛋白原纤维的二级,三级和四级结构。这些结构提供了分子堆叠的相互作用信息,有助于了解运甲状腺素蛋白从单体组装成原丝,再到成熟原纤维的过程42。此外,Jeon等2在对RSV CA的结构研究中,仅利用固体NMR所得的原子位点的扭转角信息与Cryo-EM所得结构结合,将分辨率从Cryo-EM的24 Å提高到1.5 Å。Cryo-EM擅长表征蛋白质的刚性结构域的特性,而固体NMR可同时描述刚性和柔性的结构信息。Sborgi等43在ASC炎性体结构的研究中,结合Cryo-EM的结构信息和固体NMR的扭转角信息以及柔性区域的结构信息,得到了更高分辨率的结构模型。
4 固体NMR在蛋白质组装体结构解析中的应用
4.1 Aβ淀粉样蛋白纤维结构解析
淀粉样蛋白纤维一般由正常的蛋白质异常累积形成,常伴随着蛋白质构象错误折叠。Aβ淀粉样蛋白具有神经毒性,与阿尔兹海默症(AD,又名老年痴呆症)密切相关。Aβ由淀粉样前体蛋白(APP)经过β-分泌酶和γ-分泌酶两步切割形成,常见类型有Aβ40和Aβ42。Aβ淀粉样纤维结构的解析有助于理解其聚集机制和致病机理。
运用固体NMR方法,Walti等5和Colvin等16先后发表了Aβ42的高分辨纤维结构,两个结构显示出相同的“S”型单体结构特征(如图2c),并且单体间的堆叠方式(包括垂直于纤维轴的侧向堆基和平行于纤维轴的轴向排列)也十分类似。通过STEM的暗场图像分析Aβ42纤维的MPL值,可确认单体侧向堆积数目。MPL定义为每0.47 nm长度的纤维单丝的相对分子质量:公式为MPL = M × n ÷0.47,其中M表示蛋白单体相对分子质量,n为每个β层的单体数目,相邻的β片层间距约为0.47 nm (淀粉样蛋白纤维的X-ray衍射图案表明其β层间主链间距约为0.47 nm)。其中烟草花叶病毒(TMV)有固定MPL值(131 kDa·nm−1),常作为内标用于质量校准。Colvin等16分析表明Aβ42纤维侧向堆积为二聚体形式,如图3a。
不同标记方式可帮助区分单体内/单体间距离约束条件,如图3b。对于13C/15N均匀(homogeneous)记样品,同时出现单体内和单体间相关信号,天然丰度稀释(diluted)样品(天然丰度:13C/15N标记 =3 : 1)中主要出现单体内相关信号,13C :15N = 1 : 1混合(mixed)标记样品其13C-15N相关信号主要来源分子间相关。此外Loquet等44利用1-13C葡萄糖和2-13C葡萄糖标记的互补性区分单体内/单体间信号。单体内距离约束确定Aβ42单体的“S”型结构特征;分子间距离约束如L17Cγ–M35Cβ,Q15Cγ–M35Cβ等则反应出侧向排列分子间的界面信息,如图3c;分子间距离约束如S8Cα–S8Cα,L17Cδ1–L17Cγ,S26Cα–S26Cβ,V40Cγ2–V40Cβ等说明其轴向排列方式为平行对准方式。
结合STEM所得的二聚体结构信息和固体NMR所收集的497个距离约束,利用CYANA最终确定Aβ42纤维高分辨结构,主链原子的RMSD(Root-mean-square Deviation)为(0.71 ± 0.12) Å,如图2c所示。

图4 多种Aβ40纤维结构示意图Fig. 4 The various structure models of Aβ40 fibrils.
随着Aβ42和Aβ40结构研究的报道日益增多,Aβ纤维结构展现出多态性。Aβ42的结构中Colvin等16,Wälti等5和Xiao等45都发现单体内K28与C端残基存在盐桥,这对结构有重要的稳定作用;单体沿纤维轴平行对准排列,如图2c。此外Luhrs等15在2005年发表的Aβ42结构中发现不同单体的β片层交错排列,即单体n的β1和单体n−1的β2排列,形成独特的结构形态,如图2b。Aβ40的结构同样存在多态性。Petkova等46在2002年发表的结构中,Aβ40以二聚形式存在,两单体C端平行排列,如图4a。Petkova等47在2006年和Bertini等48在2011提出结构中,单体的C端反平行排列,如图4b。已报道的Osaka突变体(E22Δ)17显示出独特的结构特点,如图2d。Paravastu等49在2008年提出的结构中,Aβ40以三聚体形式存在,如图4c。此外我们课题组19在2014年提出了Aβ40在磷脂囊泡环境中形成的纤维结构,与溶液环境中形成的Aβ40纤维对比,前者纤维疏水内核更短,且三维结构也有明显不同。这些结构差异表明了磷脂膜对Aβ纤维结构的影响,对探索细胞膜环境中的Aβ淀粉样蛋白结构具有重要意义。Aβ40结构多态性是Aβ淀粉样纤维的内在属性,这些丰富的蛋白结构可帮助我们了解在生理条件下Aβ的蛋白错误折叠机理,对理解其在AD疾病中的致病机理具有重要意义。
4.2 T3SS针状组装体结构解析
细菌III型分泌系统(T3SS)存在于多种细菌中,其用于将细菌效应蛋白从细菌传送到宿主细胞中。完整的T3SS包括膜嵌入机体(即针状体)和宿主膜中的易位孔。T3SS针状体起着细菌与细胞的通道连接作用,使细菌效应蛋白通过该通道注入宿主细胞。T3SS针状体主要由MxiH蛋白自组装形成长约100 nm,宽约8 nm的针状中空丝,单体间排列方式如图5a,b。了解针状体的结构与功能帮助我们解决细菌感染,噬菌体入侵等问题。
T3SS针状体的结构解析采用了13C的稀疏标记方法,大大提高了谱图分辨率,得到了完整的化学位移归属50;并根据二级结构特点确立了MxiH单体的螺旋发夹式折叠特征,如图5c。与Aβ42纤维类似,T3SS针状体的组装方式包括侧向堆积和轴向排列,如图5a。分别利用[1-13C]葡萄糖碳源标记,[2-13C]葡萄糖碳源标记,以及[1-13C]葡萄糖 :[2-13C]葡萄糖 = 1 : 1的混合标记样品收集结构约束条件,从中提取出明确的单体内/单体间结构约束信息,构建单体排列的结构模型,如图5c。由于单体i同时与6个其它单体接触(i ± 5、i ± 6和i ± 11,如图5a,d),界面信息复杂,存在多种约束可能性,故而结合图5c中的结构模型和固体NMR信号的距离上限(~9 Å)进行排除,如图5d。对于无法手动排除的模糊约束,在结构计算中采用迭代的算法进行自动归属。最终,从固体NMR谱中共获得996个确定的长程约束。

图5 T3SS针状体单体界面示意图Fig. 5 Intermolecular interfaces of the TSSS needle.
利用cryo-EM技术对T3SS组装体进行分析,得到分辨率为7.7 Å的结构低温密度图。为了提高T3SS针状体结构的分辨率,提出了固体NMRCryo-EM-Rosetta联用技术。固体NMR和Cryo-EM两种技术手段所得的结构信息在分辨率水平上存在差异,整合这两套数据成为实现技术联用的关键。这里利用Rosetta设计出一种迭代方案来整合这两套数据,该方案可以调整每种数据的权重大小以满足数据间的自洽性,使得两种技术手段的结构信息可以同时应用于结构计算,大大提高结构的分辨率。最后计算得到精确度高达0.4 Å的高分辨结构。固体NMR-cryo-EM-Rosetta联用方法获得了分辨率高于任何单一手段的T3SS针状结构,充分证明的技术联用的优势和可行性,可推广到其它超分子蛋白组装体的结构解析中。
6 总结与展望
在过去的20年中,固体NMR技术已被证实在蛋白质组装体结构解析中具有很大潜力。常见的蛋白质组装体如蛋白质细丝、原纤维或病毒衣壳等,它们通常不易结晶且难溶解,传统的结构解析方法如液体NMR,X射线晶体学方法等很难得到高分辨结构,而固体NMR可在原子水平对这些生物体系进行高分辨结构研究。固体NMR实验技术的提升和样品制备方法的改进都大大促进了固体NMR在蛋白质组装体研究领域的发展。采用多种13C稀疏标记和选择性标记方法有助于提高谱图分辨率,混合标记方法可区分单体内/单体间距离约束条件;多种实验方法如DARR、PDSD、PAR和TEDOR等可以有效地获得距离约束,为高分辨结构解析提供条件。氢检测方法和顺磁标记技术的发展为固体NMR获取蛋白质结构信息提供了新思路,在蛋白组装体结构的研究中具有很大潜力。固体NMR与多种研究手段(如STEM、X-ray、Cryo-EM、MD、Rosetta等)的强强联合,在解析复杂蛋白质体系结构方面展现出显著优势。随着固体NMR技术以及相关实验技术的进一步发展,固体NMR在蛋白质组装体结构解析领域发挥着越来越重要的作用,为相关生物分子的功能和致病机理提供重要的结构信息。
猜你喜欢
网络安全与数据管理(2022年1期)2022-08-29
储能科学与技术(2022年2期)2022-02-19
气象与环境学报(2021年3期)2021-07-14
矿产勘查(2020年3期)2020-12-19
家庭影院技术(2018年9期)2018-11-02
三联生活周刊(2017年48期)2017-11-25
小学阅读指南·低年级版(2017年1期)2017-03-13
人生十六七(2015年6期)2015-02-28
计算机辅助工程(2012年5期)2012-11-21
微型计算机(2009年4期)2009-12-23
