
新型磁性分子印迹电化学传感器对食品中敌草隆的检测
2020-08-06 11:43肖维玮卢一辰熊晓辉
生物加工过程 2020年4期
肖维玮,卢一辰,熊晓辉
(南京工业大学 食品与轻工学院,江苏 南京 211800)
敌草隆(diuron,DU)是一种苯基脲类除草剂,其化学名称为N-(3,4-二氯苯基)-N,N-二甲基脲,被广泛应用于小麦和水稻田中去除一年生和多年生阔叶、杂草[1]。大量研究表明,敌草隆及其代谢产物的环境持久性致使其环境残留物过量积累,对非靶标动植物产生毒性作用,甚至有研究发现敌草隆代谢产物可能对人类健康和生殖产生严重影响,浓度为5 mmol/L的敌草隆主要代谢产物3,4-DCA能够在半小时内对人类精子产生损伤[2-3],可见对环境和食品全链条的敌草隆残留物进行检测和监控势在必行。近年来,已陆续建立了高效液相色谱法(HPLC)、液相色谱-质谱法(LC-MS)、气相色谱法(GC)等测定环境样品中敌草隆的分析方法。然而,这些方法受仪器、高检测成本以及前处理步骤繁琐的限制,已经不能满足现阶段大量样品的快速检测筛查需求[4]。因而,开发一种简单、灵敏、廉价、快速的分离检测环境或食品介质中的敌草隆分析方法具有重要意义。
1972年,Wulff等[5]首次报道通过共价法合成了分子印迹聚合物(MIPs),因其具有靶标化合物契合的空穴结构,对目标物具有类似于“抗体-抗原”的特异结合能力,开始引起学术界的关注。以茶碱为模板,采用非共价法合成的印迹材料在《Nature》上发表后,在学术圈引起了巨大反响[6],其解决了共价法引起的模板不易洗脱的难题,使得分子印迹材料成为研究热门。刘娇等[7]使用本体聚合方法制备了能够对甲基对硫磷进行特异性吸附的分子印迹材料,其饱和吸附量可达625.5 μmol/g,可用于水样中甲基对硫磷的富集与分析。田景升等[8]使用表面分子印迹技术,以Fe3O4@SiO2为载体、甲基丙烯酸酯为功能单体、联苯三唑醇为模板分子,制备了联苯三唑醇分子印迹聚合物。Uygun等[9]在石墨电极上电聚合吡咯,制备了能够对毒死蜱特异性吸附的分子印迹膜,其制备的传感器具有成本低、选择性优良、灵敏度高等特点。分子印迹聚合物对目标物具有较高的选择性,且物理性质、化学性质相对稳定,制备过程简单,易保存。基于上述特性,分子印迹材料作为优良的前处理材料大量应用于食品和环境样品之中,例如:固相萃取和固相微萃取材料[10-11]、色谱固定相材料[12]以及分子印迹传感器[13]等。
近年来,以分子印迹聚合物为识别元件,结合不同类型的转换器而制得的分子印迹传感器,其兼具生物传感器的专一识别性和化学传感器的力学稳定性、热稳定性等优点,成为传感器研究领域的新热点。电化学传感器的高超灵敏性易受到基质效应的影响,导致结果重现性较差。为了改善这一问题,许多研究者将具有选择性识别位点的分子印迹材料与电化学传感器相结合[14],来达到同步专一性识别和检测目标物的目的。传统的电化学MIPs传感器采用电聚合修饰的分子印迹膜,面临着电极修饰步骤繁琐、刷新电极表面耗时长等问题,限制了分子印迹电化学传感器的进一步应用和发展[15-16]。因此,开发更为便捷的整合方式尤为重要。
磁性分子印迹材料的开发给打破繁琐电极修饰带来了灵感。磁性分子印迹是以顺磁性材料为核,在其表面聚合分子印迹层,在不影响其磁性的情况下通过添加一个外部磁场,使磁性分子印迹特异性结合目标物后可以很容易地从样品中分离出来,避免了耗时的离心步骤。这样一个可控的重新绑定过程能够简单、快速地进行磁分离操作。因此,若利用此类分子印迹材料的磁吸附性能和特异性吸附作用,有望简化电极表面修饰过程和提高电化学传感器的专一性,同时可消除传统电极系统中再生困难等问题。
因此,本文中,笔者以敌草隆为模板分子,采用表面聚合方式在磁性Fe3O4纳米粒子表面合成印迹材料,并通过磁吸附作用构建一种新型的磁性分子印迹电化学传感器用于测定食品介质中敌草隆的含量。
1 材料与方法
1.1 材料和仪器
敌草隆,远成共创科技有限公司;甲醇、乙腈、无水MgSO4、亚铁氰化钾、铁氰化钾、KCl(分析纯),广东光华科技有限股份公司;甲基丙烯酸(MAA)、三羟甲基丙烷三甲基丙烯酸酯(TRIM)、3-(甲基丙烯酰氧)丙基三甲氧基硅烷(MPS)、正硅酸乙酯(TEOS)、氨水、异丙醇、甲基丙烯酸、偶氮二异丁氰(AIBN)、柠檬酸三钠、Fe3O4(分析纯),阿拉丁有限公司;磁性玻碳电极(Φ=5 mm)、AgCl电极、铂丝电极,天津英科联合有限公司。
电子分析天平,上海天平仪器厂;组合式摇床,太仓市强乐实验设备有限公司;数控超声波清洗器,昆山禾创超声仪器有限公司;超纯水器,北京普析通用仪器有限公司;真空干燥箱,上海精宏设备有限公司;CHI750E型电化学工作站,上海辰华科技有限公司。
1.2 实验方法
1.2.1 磁性分子印迹材料合成
敌草隆磁性分子印迹材料参照文献[17]方法。
首先,使用柠檬酸三钠改性Fe3O4,称取2.0 g Fe3O4纳米粒子与0.1 mmol/L的柠檬酸三钠溶液混合超声30 min,40 ℃ 条件下机械搅拌24 h(通氮气)。待反应结束后,离心收集产物,用超纯水、丙酮清洗若干次,真空干燥保存备用。接着用TEOS对改性Fe3O4进行修饰,称取1.0 g改性Fe3O4纳米粒子分散在混有20 mL超纯水、4 mL氨水(质量分数32%)和120 mL 2-丙醇的溶液中。超声处理30 min后,缓慢滴入含有20 mL的2-丙醇和6 mL的TEOS混合溶液中,机械搅拌20 h(通氮气),用磁铁收集Fe3O4@SiO2产物,并用超纯水和乙醇清洗若干次,真空干燥保存。
然后,用3-(甲基丙烯酰氧)丙基三甲氧基硅烷(MPS)对制备的Fe3O4@SiO2进行修饰,称取300 mg Fe3O4@SiO2分散在90 mL甲醇溶液中,通N2机械搅拌5 min后缓慢滴入5 mL MPS-甲醇溶液(体积比为1∶ 4),接着在30 ℃条件下继续搅拌24 h。使用磁铁收集产物,并用甲醇清洗若干次,真空干燥保存。
最后,取0.625 mmol/L敌草隆和2.5 mmol/L甲基丙烯酸(MAA)溶解在乙腈溶液中,冰浴条件下搅拌12 h进行预聚合反应。在上述溶液中添加300 mg的Fe3O4@SiO2@MPS,N2保护下搅拌3 h后,将9 mmol/L 三羟甲基丙烷三甲基丙烯酸酯、50 mg偶氮二异丁腈溶于乙腈后加入上述反应物中引发聚合。首先在50 ℃下搅拌反应6 h,接着在60 ℃下搅拌反应24 h(全程N2保护)。在不存在模板分子敌草隆的条件下,其他相关的合成条件与敌草隆印迹聚合物(DUMIPs)相同,制备敌草隆非印迹聚合物(DUNIPs)。合成反应结束后,以甲醇-醋酸溶液(体积比9∶ 1)为洗脱液,使用索氏提取法在110 ℃条件下对聚合物进行洗脱,在250 nm处检测洗脱液的浓度,直至洗脱液中无法检测出敌草隆,真空干燥保存。
此外,随着全球气候变化,极端干旱、洪涝灾害事件在增加。应对气候变化对灌排工程的改造和发展也提出了更高的要求。水利设施是提高农田生产效率的重要基础设施。今后粮食的增长和解决灌溉水资源短缺的问题必须依靠提高灌溉用水效率和效益。但从现实来看,缺乏灌排设施,以及已有灌排设施的老化是发展中国家和最不发达国家面临的一个较为普遍的问题。面对这些问题,一些国家缺乏在国家层面上的政策支持,缺乏资金和技术,无力开展灌排工程设施的改造和升级。
1.2.2 电化学传感器的制备
将磁性玻碳电极放在加有泥状Al2O3(0.05 μm)的麂皮上进行抛光打磨,依次放入无水乙醇和超纯水中超声清洗以除去电极表面的Al2O3粉末及其他杂质;在-0.2~1.2 V的电位下,将磁性玻碳电极放置在0.5 mol/L的H2SO4溶液中进行循环伏安法的扫描,直至得到较为稳定的循环伏安曲线。将3 mg的磁性分子印迹聚合物放入10 mL不同浓度的敌草隆溶液中,摇床吸附30 min,使磁性MIPs与敌草隆充分结合。5 min内将磁性玻碳电极插入敌草隆溶液中,通过磁力作用将溶液中已吸附完全的印迹材料固定到电极表面。
1.2.3 电化学传感器的测定
以50 mL含有0.1 mol/L KCl的2.5 mmol/L的Fe(CN)63-/4-作为电解液,磁性玻碳电极作为工作电极,AgCl电极作为参比电极,铂丝电极作为对电极,用循环伏安法(CV)和示差脉冲伏安法(DPV)对修饰电极和裸电极进行了表征。循环伏安(CV)模式下扫描范围为0~0.55 V,扫描速率为100 mV/s。示差脉冲测量(DPV)模式下,脉冲宽度为50 ms,脉冲振幅为25 mV,脉冲周期为50 ms,电位增量为5 mV。所有测量均在室温条件下进行,且最终靶标检测的电化学方法与上述相同。
1.2.4 实际样品制备
取2 mg MIP加入到5 mL含不同浓度(0.2、1、4 mg/L)的敌草隆自来水样品中,25 ℃下振荡吸附30 min。接着将磁性玻碳电极插入到电解液中2 min,通过磁吸附作用,使得溶液中的MIP固定到玻碳电极表面。茶饮料样品的制备过程与此相同。
取5 g大白菜切片(约1 cm×1 cm),加入5 mL不同浓度的敌草隆,静置30 min后加入10 mL甲醇到样品中,超声20 min。然后将混合物8 000 r/min离心10 min。接着在50 ℃的旋转蒸发器中使上清液挥发干燥,用200 μL甲醇溶解残余敌草隆,并用5 mL超纯水稀释。吸附过程与水样相同。
2 结果与讨论
2.1 分子印迹的合成与电化学的检测原理
图1展示了磁性分子印迹的合成和电化学的检测过程。

图1 磁性分子印迹的合成和电化学的检测
如图1所示,所合成的磁性分子印迹材料以Fe3O4为内核,使用柠檬酸三纳对其改性,增加其分散性,接着使用TEOS对柠檬酸盐改性的Fe3O4进行修饰,在其表面包裹了一层SiO2阻碍磁珠的团聚和氧化。接着使用MPS对Fe3O4@SiO2进行修饰,双键的接枝便于后续聚合反应的进行。然后,以DU为模板分子、MAA为功能单体,TRIM为交联剂,AIBN为引发剂,通过自由基引发聚合,在MPS-Fe3O4@SiO2表面合成敌草隆印迹聚合物。使用磁性玻碳电极,通过磁力作用,将吸附了敌草隆的磁性分子印迹材料固定到玻碳电极表面,在含有0.1 mol/L KCl的2.5 mmol/L的Fe(CN)63-/4-溶液中进行脉冲伏安法的检测,导电的敌草隆分子增强了电解液中电子的传递,因此,随着敌草隆浓度的增高,所检测循环伏安法的电流值越大。
2.2 敌草隆导电性的验证
为了考察敌草隆的导电性,将裸电极分别放入空白溶液和敌草隆溶液中,通过CV法观察其信号的变化。图2为玻碳电极检测空白和不同浓度敌草隆溶液的CV图谱。
由图2可知,玻碳电极在敌草隆溶液中的氧化还原信号高于空白溶液。同时,随着敌草隆的浓度从0.000 1增大到0.001 mmol/L,氧化峰电流和还原峰电流逐渐增大。因此,说明敌草隆是导电物质。导电的敌草隆分子可以促进Fe(CN)63-/4-发生氧化还原反应,使得溶液中的电子通过印迹空穴转移到磁性玻碳电极表面,因此,随着敌草隆浓度的增加,靶标电信号也随之提升。

图2 空白和0.000 1 mmol/L DU、0.001 mmol/L DU溶液在MGCE上的CV信号
2.3 磁性分子印迹材料添加量的优化
电极表面的MIPs量是影响电信号传导的重要因素。因此,为了优化MIPs的添加量,将吸附不同量敌草隆的印迹材料(DUMIPs)与未吸附不同量敌草隆的印迹材料(CKMIPs)负载到玻碳电极表面,测量其电信号的变化,结果如图3所示。

图3 敌草隆印迹材料在磁性玻碳电极(MGCE)上添加量的影响
由图3可知,负载在玻碳电极表面MIPs的量对峰电流影响较大。随着DUMIPs与CKMIPs的量从0.5 mg增加到2 mg,峰值电流逐渐降低,电流下降的原因可能是由于不导电的MIPs层阻碍了电子传递[17]。接着增加DUMIPs与CKMIPs的量,DUMIPs的电流继续下降;然而,当CKMIPs的量达到2 mg时,峰值电流急剧增加,其增加的原因是CKMIPs的量太多,导致非导电的MIPs层在电解质溶液中脱落,因此,CKMIPs的峰值电流出现了先下降后上升的波动。当MIPs的量为2 mg时,DUMIPs和CKMIPs之间的峰值电流差异最明显。同时,2 mg的MIPs足以完全覆盖MGCE表面,不会在电解质溶液中脱落。因此,使用2 mg MIPs修饰MGCE表面最佳。
2.4 MIP在MGCE表面负载时间的优化
图4显示了负载时间对MIPs在MGCE上累积后产生的DPV电流响应的影响。由图4可知,在5~30 min时,峰值电流随MIPs负载时间的变化不明显,说明短时间内DUMIPs通过磁性作用能很快与MGCE结合。因此,选择5 min进行进一步的实验,可大大节省负载时间。

图4 DUMIPs在玻碳电极表面负载时间的影响
2.5 MIPs、NIPs、Fe3O4@SiO2结合敌草隆效果的验证
为了考察分子印迹技术和磁性富集技术对敌草隆的吸附效果,进行了验证实验,结果如图5所示。

a、b表明MIPs分别与NIP、Fe3O4@SiO2具有显著性差异和非显著性差异
由图5可知,NIPs传感器与Fe3O4@SiO2传感器在吸附敌草隆模板前后,电流值并没有明显的改变。这是因为NIPs传感器和Fe3O4@SiO2传感器没有与敌草隆相匹配的空腔。而MIPs在吸附前后,其电流值呈现出一个较为明显的改变,ΔIp=26.13 μA。这种显著性差异表明MIPs选择性位点的高效性。敌草隆位点位于印迹材料表面,方便吸附溶液中的敌草隆模板分子。由此表明分子印迹技术和磁性富集技术可大大提高该电化学传感器的特异性。
2.6 电化学行为表征
图6为Fe(CN)63-/4-溶液中裸电极和修饰电极的循环伏安图。扫描速率为100 mV/s,扫描范围为0~0.55 V。
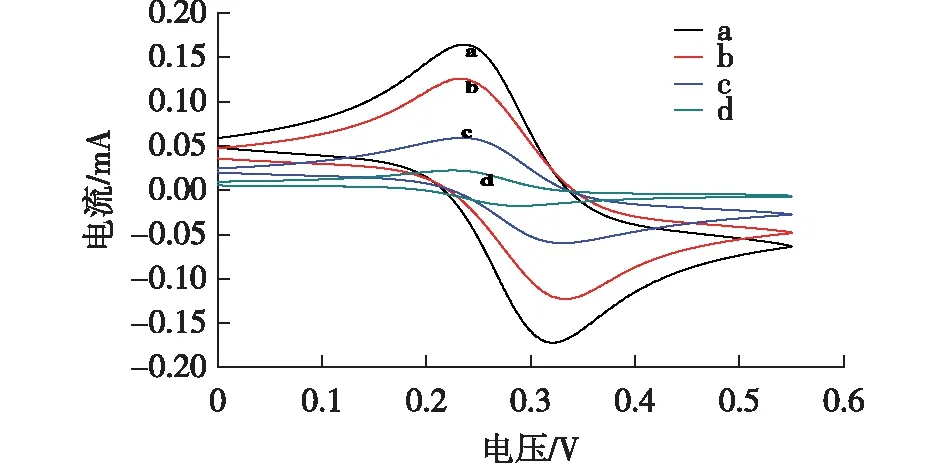
图6 裸MGCE(a)、模板去除前的MIPs(b)和模板去除后的MIPs(c)、非印迹材料(NIPs)(d)的循环伏安曲线(CV)
由图6可知,曲线a显示出一对明显的裸电极的氧化还原峰,峰值电流为161.34 μA,峰值电位(△Ep)为101.12 mV。电极在被NIPs覆盖后,未出现明显的氧化还原峰(曲线d),这是由于NIPs在聚合反应时没有敌草隆模板,印迹材料并没有形成能够对敌草隆进行特异性吸附的空穴,从而阻止了电解液从溶液中转移到电极表面[18]。曲线c为敌草隆模板分子去除后的曲线,与NIPs/MGCE相比,氧化还原峰值电流增加,由于NIPs的合成并没有导电敌草隆分子的参与,因此NIPs也不存在能够对敌草隆进行特异性吸附的空穴,而MIPs在洗脱敌草隆分子之后,会留下特异性空腔,洗脱之后的空腔增强了Fe(CN)63-/4-通过MIPs层的扩散,促进了Fe(CN)63-/4-在传感器上的氧化还原反应[19],但是,它的增强效率远低于结合敌草隆分子的MIPs。曲线b为敌草隆模板分子去除前的曲线,与去除敌草隆模板后的MIPs电流相比,电流明显增大,可能是残留在空腔中的敌草隆促进了Fe(CN)63-/4-通过MIPs层的扩散。
2.7 标准曲线
在最优实验条件下,将制备好的MIPs分散在不同浓度的敌草隆溶液中,吸附5 min,结果如图7所示。
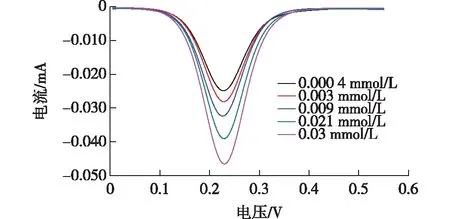
图7 DU的差分脉冲伏安法(DPV)信号
由图7可知,检测到的DPV信号随着敌草隆浓度的增加而增加,这是因为当敌草隆分子进入分子印迹层的空穴时,促进了Fe(CN)63-/4-溶液中的电子转移到磁性玻碳电极表面,改善了电化学响应。敌草隆浓度在0.000 4~0.03 mmol/L范围内有着较好的线性关系,线性方程为y=0.724 9x+0.024 7(R2=0.992 9),且最低检测限(LOD)为1.3×10-5mmol/L(S/N=3)。
2.8 实际样品检测
为了评价印迹传感器的实际应用性,在自来水、茶饮料和大白菜样品中进行加标回收实验。向上述实际样品分别直接添加质量浓度为0.2、1、4 mg/L的敌草隆,采用优化后的前处理和检测方法测定实际样品中的敌草隆含量。检测的回收率和相对标准偏差如表1所示。
由表1可知:敌草隆在3种实际样品中的回收率分别为95.0%~101.5%、101.6%~110.0% 和100.5%~105.5%,且RSD值均小于9.6%。样品的良好回收率和较低的RSD值表明该传感器对快速测定敌草隆有很好的实际应用价值。

表1 实际样品加标回收率(n=3)
3 结论
以Fe3O4为内核、SiO2为外壳所制备的磁性分子印迹聚合物,通过磁吸附作用使其固定到磁性玻碳电极表面,成功构建了能够定量检测敌草隆的分子印迹电化学传感器。敌草隆浓度在0.000 4~0.03 mmol/L内可被定量检测,检测限(LOD)为1.3×10-5mmol/L。该方法成功用于不同实际样品的敌草隆测定,验证了此传感器用于真实样品分析的能力。
所构建的磁性分子印迹电化学传感器有如下优点:①将分子印迹技术与磁性纳米粒子相结合,可以实现可控的重结合过程和简单快速的磁分离;②磁性玻碳电极能够实现多样品检测,电极再生简单;③成本低、响应快、操作简单,可实现实时检测。因此,本文中笔者所构建的磁性分子印迹电化学传感器为敌草隆的检测创造了一个良好的平台,同时在食品分析与环境监测中具有潜在的应用价值。
猜你喜欢
陶瓷研究(2022年3期)2022-08-19
云南画报(2021年10期)2021-11-24
中学生数理化(高中版.高考理化)(2021年4期)2021-07-19
中学生数理化(高中版.高考理化)(2020年10期)2020-10-27
陶瓷学报(2019年6期)2019-10-27
表面工程与再制造(2019年6期)2019-08-24
发明与创新·小学生(2019年11期)2019-08-11
小学生优秀作文(高年级)(2018年4期)2018-09-11
资源节约与环保(2018年1期)2018-02-08
军事文摘·科学少年(2017年4期)2017-06-20
