
神经网络全局势函数在多相催化中的应用
2020-09-23 09:31马思聪刘智攀
化工进展 2020年9期
马思聪,刘智攀
(复旦大学化学系,上海200433)
自20 世纪90 年代开始,随着计算机技术的发展和量子力学的深入研究,多相催化理论计算逐渐成长为一门独立的学科,为催化领域的发展添砖加瓦。理论催化的最大优势在于其研究的角度是从原子和分子层面出发的:构建催化剂结构模型、研究反应物分子的吸脱附行为、探索不同反应路径来确定化学反应动力学,最终构建出催化剂结构与催化性能之间的关系。
“构建催化剂结构模型”寥寥几个字背后是一片高度复杂的荆棘丛林。材料结构的模拟和预测是理论化学研究中一大重要方向,只有熟知材料结构才能有的放矢,实现后续的化学反应研究。材料结构模拟的难点在于其复杂度上,N个原子的体系有3N 个坐标,不同的坐标可以产生无穷种可能的结构,如何搜索到热力学合理的结构一直是这一领域的难点。目前已发展出很多比较成熟的势能面采样方法,包括随机表面行走(stochastic surface walking,SSW)、遗传算法(genetic algorithm)、盆地跳跃(basin-hopping)等(具体在第1 节详细介绍)。在实际应用中,势能面搜索通常都是基于第一性原理方法来计算能量和力。虽然该方法得到的势能面比较准确,但本身的计算速度太慢,获得一个结构的能量和力的计算代价过大。因此,为了更快地遍历势能面,可以从改变能量计算方法角度来着手。目前,比较有前途的措施就是把第一性原理方法改成力场方法。采用力场方法可以实现对势能面的快速搜索,比第一性原理方法快103~105 倍,但力场方法的精确度一直备受诟病。如何构造出实用的力场一直是目前的研究重点。在构造力场的方法中,神经网络(NN)是一种强大的拟合工具,可以根据已有数据表达能量和结构之间的函数关系,从而实现预测功能。既获得了计算精度,又可以实现计算速度的提高。目前,本文作者课题组开发了LASP(large-scale atomic simulation with neural network potential)软件用来解决相关的技术问题[1]。LASP 核心优势在于:①可以利用神经网络进行势能面模拟计算;②提供了SSW 势能面采样工具;③提供了高效的双端表面行走方法(double-ended surface walking method,DESW)过渡态搜索工具。
1 SSW-NN方法介绍
1.1 势能面结构搜索方法介绍
在Born-Oppenheimer 近似下,整个体系的能量可以看成与原子核坐标相关的函数。N个原子的体系具有3N个自由度,体系的能量可以看成3N维空间的超曲面,这个超曲面叫作势能面。图1显示了一个三维投影的势能面示意图。这个势能面有几个需要注意的地方:首先是能量的局域极小点,这几个点的一阶梯度为0,Hessian 矩阵(二阶梯度)的本征值全部为正值。这些点一般都是代表着稳定结构,有可能在实际中被合成出来。其次是几个鞍点,这几个鞍点的一阶梯度为0,但是Hessian 矩阵的本征值中有一个为负值,意味着在这个方向它是不稳定的,这些鞍点也被称为过渡态。对于化学反应研究,势能面采样的核心就是如何快速得到极小点和鞍点。

图1 三维势能面示意图
假设有一个结构A,这个结构并不是极小值点。此时,为得到A结构附近的极小值点结构,需要对结构A进行能量极小化,这个过程叫作结构优化。由于每个极小值点就是一个势能面上的“坑”,所以结构优化更通俗地讲叫作“入坑”。“入坑”的方法有很多,包括共轭梯度算法(conjugate gradient)、牛顿法(Newton method)、准牛顿法(quasi-Newton)和Broyden 方法等。“入坑”的方法只适用于从一个点出发搜索局域稳定结构,对全局势能面搜索则没有助益。因此,必须要有“出坑”的方法来扩大搜索范围。目前在“出坑”方面已开发出大量方法。盆地跳跃算法[2-3]是基于在某一随机方向上添加微扰,然后进行结构优化以及最后通过蒙特卡洛(Monte Carlo)选择过程来确定是否接受新结构。基本步骤为从当前结构出发,在某一方向上随机移动原子,并对移动后的结构进行几何结构优化,然后根据一定标准来决定是否接受新结构。整个算法就相当于是在一个个盆地里面来回跳跃。元反应动力学(metadynamics)算法[4-5]是在分子动力学的基础上,往势能面上添加偏置势函数来变相降低跳跃能垒,加快翻越势能面的速度。遗传算法[6-7]是选取一系列结构(亲代),通过结构之间的相互组合、交叉、变异从而产生出新的结构(子代)。再根据能量筛除高能量结构,保留低能量结构。通过反复的迭代,实现势能面结构遍历和搜索。粒子群优化(particle swarm optimization)算法[8-9]则采用一群结构,对这群结构给一个向着能量最低结构的微扰方向来产生新结构。模拟退火(simulated annealing)[10-11]则是沿着在某个方向行走并产生一个不断升降温的蒙特卡洛选择过程来确定是否接受新结构。极小值跳跃(minima hopping method)[12]采用分子动力学方法,在相同能量的极小值结构附近不断升温,使原子位置偏离极小值结构从而产生新结构。本文作者课题组发展了SSW方法来搜索势能面[13-14],主要借鉴了元反应动力学方法中添加偏置势函数的思想,不断添加势函数使其可以翻山越岭。在各类算法中,有些侧重于寻找新结构,有些侧重于寻找结构之间的相变路径。一般来说,这两种是互斥的。道理很简单,越倾向搜索产生新结构的方法,其对原有结构的依赖越小,可以随意地变化坐标位置,也就无从谈结构之间的关联性。而寻找结构之间关系的方法,则对原有结构非常依赖,搜索范围也就局限在原有结构附近,搜索效率降低。
1.2 SSW方法
SSW 方法是基于偏置势函数驱动分子动力学的思想发展而来的。整个SSW过程包含三个步骤,即爬山步骤、优化步骤和蒙特卡洛判断步骤,其中核心是第一个爬山步骤。具体来说,首先从一个稳定的结构点出发,在某一随机方向添加偏置势函数。添加的高斯势函数用式(1)来表示。

式中,NG 为添加高斯势函数的个数;R 是结构的坐标向量;Vreal是真实的势能面;Rtn是添加n次高斯势函数以后的结构坐标向量。高斯势函数的大小和宽窄用w 和ds 来控制。然后优化这个方向,使得这个方向的振动频率比较低。继续随机添加偏置势函数,优化方向。反复迭代到一定次数后,撤去偏置势函数,利用“入坑”方法优化到底,最后通过蒙特卡洛过程来决定是否接受新结构,见图2。真实的势能面用黑色实线表示,从一个局域极小值到另一个局域极小值是通过一步一步在某个方向上添加高斯势函数实现的。目前SSW 方法已经可以实现晶体、 团簇、 分子和表面结构搜索[13,15-17]。
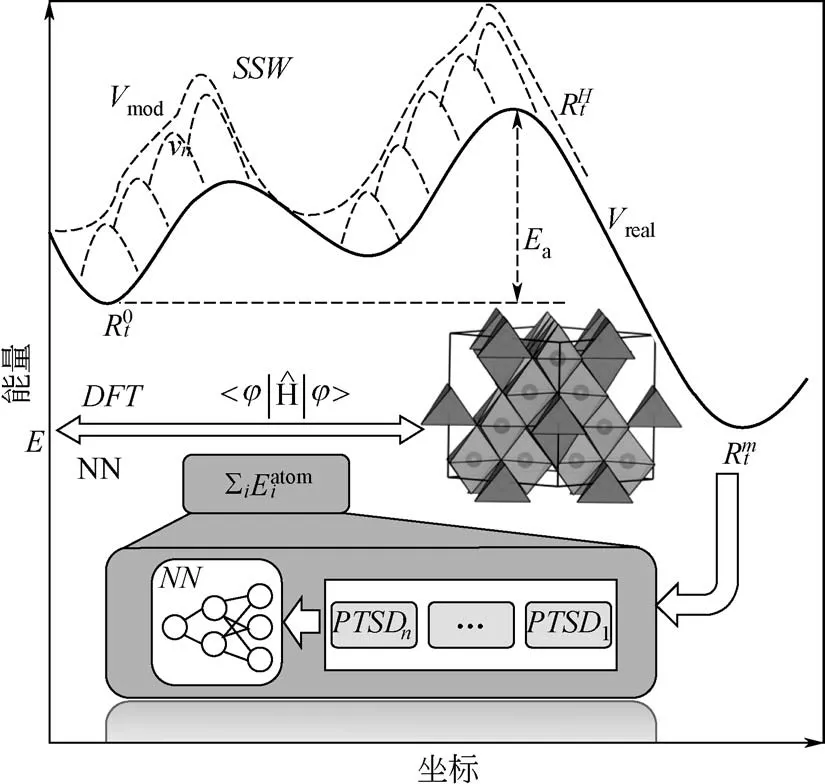
图2 SSW-NN示意图
1.3 NN方法
1.3.1 NN架构
神经网络作为一种强大的拟合算法,近年来为催化领域带来了新的辉煌[18]。它最初是用来研究大脑中的信号处理[19]。在接下来的几十年里,神经网络已经发展成为一类广泛的算法,在数字预测、模式识别和数据分类等领域有着广泛的应用。通过非线性“黑盒”数据处理,自动搜索独立或非独立变量与目标值之间的函数关系。目前,人们已经尝试利用神经网络来预测化学性质,例如振动光谱[20]、力常数[21]、两分子的反应散射、单分子分解反应和气体分子在表面上的吸附等[22-23]。因此,神经网络在搜索结构和解决反应活性位点方面显示出巨大的潜力。
图3显示了一个最简单的Feed-Forward 神经网络(FFNN)的工作原理。整个网络由许多神经元或按层排列的节点组成。输入层中的节点输入所有可提供的信息(xi)。输出层节点的最终值(y)是xi的一个解析函数。而函数形式则由隐藏层的数量和每个隐藏层的神经元数量决定。图3采用线性方程组形式和矩阵形式列出了数据是如何在FFNN传递的。值得注意的是,每一个获得的值(yij)都应该被一个非线性活化函数来活化。神经网络的核心是获得在基于梯度的迭代优化算法下确定的最优参数(wikj)。一个好的神经网络势函数受许多因素的影响,例如隐藏层的数量、每个隐藏层的神经元数量、活化函数和优化算法。在实际应用中,采用几种不同的网络结构来构造神经网络,分析结果,选择最佳网络结构是最有效的方法。在早期,FFNN结构被引入化学领域。输入层中的节点以坐标向量(如笛卡尔坐标或带距离和角度的内部坐标)的形式来描述化学结构,输出层是总能量。这就要求训练集必须具有相同数量的原子,才能在神经网络势函数中保持相同的输入层节点数。所得到的神经网络势函数也只能用于原子数与训练集相同的系统,导致FFNN势函数没有实际的预测价值。
2007年,Behler和Parrinello首先实现了高维神经网络框架(HDNN)以满足实际应用,该框架可适用于含有数千个原子的高维系统(图3)[24-25]。在这种框架中,通过拆分,使每种元素具有一个FFNN,一个体系中的每个原子分别进入各自对应元素的FFNN来得到目标值。通过这种构想可以克服早期FFNN势函数不可扩展的问题。每个原子可以提供一个原子能量Ei,总能量便可写成所有原子能量的加和,见式(2)。

HDNN框架适用于任意数量的原子:如果一个原子从系统中被移除,它所对应的原子神经网络就会被删除;反之亦然。具体的详细过程见图3和其他参考文献[26-28]。HDNN 和FFNN 最大的区别是输出层,其中原子能和总能量分别是HDNN 和FFNN的目标值。HDNN的困难在于如何获得原子能。幸运的是,真正的原子能不需要精确计算,HDNN可以自动地将总能量拆分成“假”的原子能。因此,HDNN中最重要的是如何描述输入节点中的原子环境。如Behler等[24]所建议的,用于描述原子环境的输入节点通过解耦将局部环境中原子的笛卡尔坐标转换成特殊类型的多体结构描述符(Gi)。这些结构描述符提供了系统中每个原子的相邻原子的径向和角度排列信息。一个好的结构描述符必须是一个单值函数,每个不同的原子环境对应不同的值,并且它们必须是连续和可微的,以便能够计算力的解析导数。
原子力可通过式(3)来计算得到[29]。
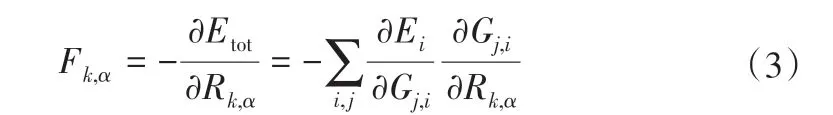
式中,Fk,α是作用在k 原子上在α(α=x,y,z)方向上的力;Rk,,α为原子坐标。结合式(2)可以得到Fk,α与Gi之间的关系。
类似地,静态应力张量矩阵元σαβ可以由式(4)得到。

式中,rd和rd分别为距离向量和它的模;V 是晶体结构的体积。
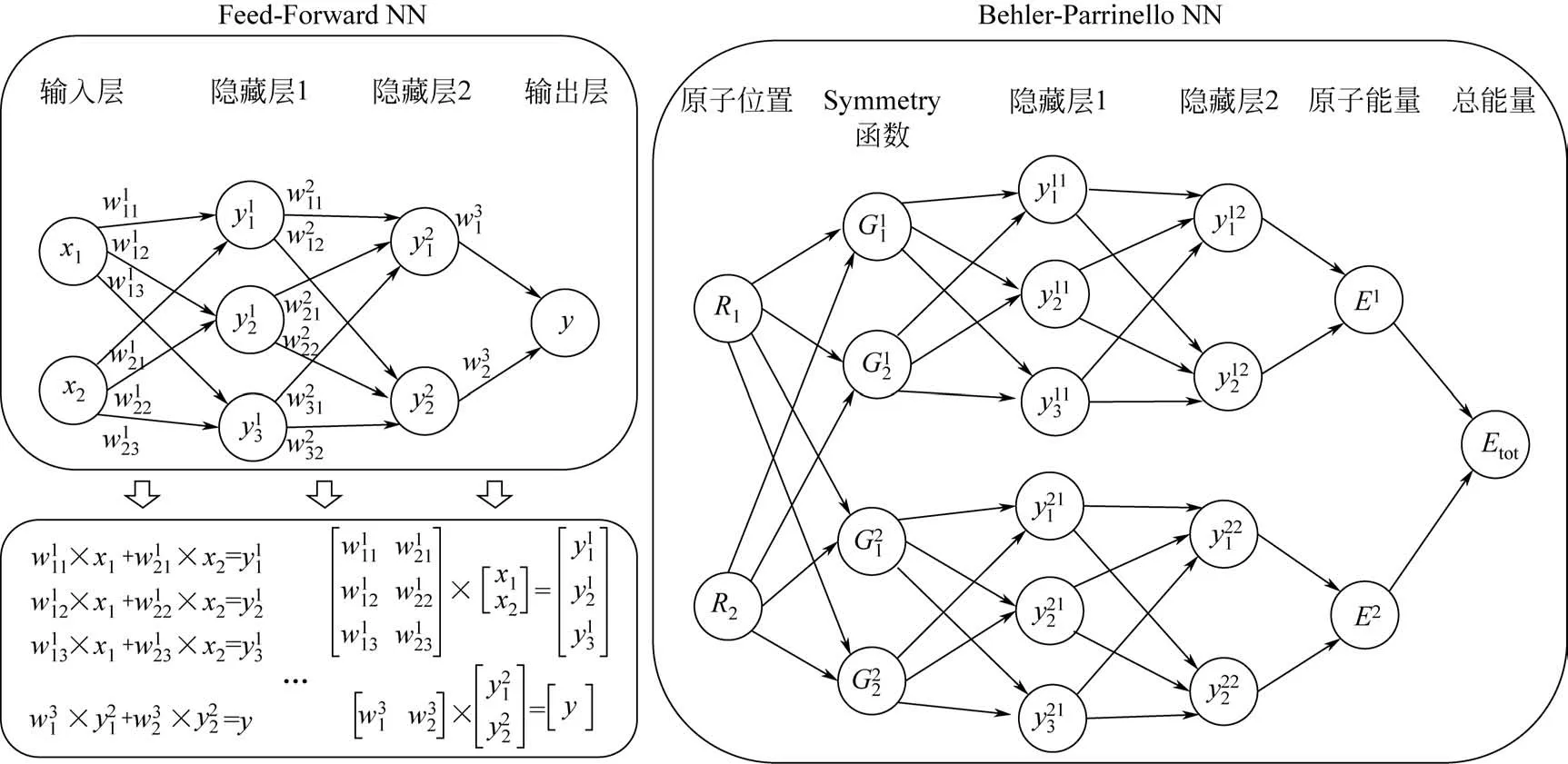
图3 Feed-Forward和Behler-Parrinello神经网络框架
一旦确定了网络架构,下一步就是确定每个NN 子网中的权重和偏差(NN 参数),这个过程称为NN训练。对于具有两个隐藏层的标准FFNN,每个隐藏层有40 个节点,那么权重和偏差的数量通常为104~106。为了训练如此大量的神经网络参数,需要定义一个性能函数Jtot[29-30]来测量神经网络输出相对于训练集真实值之间的偏差,见式(5)。训练过程将使Jtot最小化,直到神经网络预测属性的精度达到预设标准。

式中,ρ=1~100和τ=0.1~1。这允许Jtot可以同时训练能量、力和张力。在实践中发现,对于固体的全局优化,力和应力都需要精确,最方便的方法是允许神经网络训练同时或独立地调节式(5)中的三项参数。
目前许多基于梯度的优化算法已经被用于优化网络权重和偏差,如随机梯度下降(SGD)[31]、共轭梯度(CG)[32-33]、Levenberg-Marquardt(LM)[34]等。一般认为,准牛顿二阶方法,如L-BFGS和LM会更快地收敛到真正的极小值。值得一提的是,与使用量子力学计算生成数据集的工作量相比,神经网络的训练实际上不是整个过程的速度决定步骤。
1.3.2 结构描述符
在原子的笛卡儿坐标到结构描述符的转换过程中,必然要在某一半径处做截断,导致结构信息的丢失,因此应通过选择合适的结构描述符来小心地减小这种误差。作为在HDNN中将结构与其能量相关联的关键,要求一个合格的结构描述符必须要足够敏感,以尽可能地区分势能面上的每个结构。Behler 和Parrinello[24]提出了高斯型结构描述符(GTSD),式(6)~式(8)中描述了最常用的两体G2 和三体G4函数。

式中,rij是原子i 和j 之间的距离;θijk是以i 原子为中心的j 和k 为邻居形成的夹角(i、j、k 是原子指数)。GTSD中的关键成分是截断函数fc,它在rc之外衰减到零。高斯型径向函数和角向三角函数可以通过改变rc、rs、η、ζ 和λ 五个参数来产生一组G2和G4函数,用来区分中心原子i的原子环境。
然而,“exp”自然指数函数的计算速度很大程度上取决于计算机服务器类型,需要专门的计算机指令集来获得最快的计算速度,如avx512er。通常,幂指数函数的计算速度比自然指数函数快得多。因此,幂指数函数形式的结构描述符(PTSD)被提出来描述原子环境,见式(9)~式(14)[30]。

在PTSD 中,S1 和S2 是二体函数,S3、S4 和S5是三体函数,S6是四体函数。用PTSD中的幂函数代替GTSD中的高斯函数有几个优点:①减少了数值计算中的计算成本;②将可调参数从两个(rs,η)减少到一个(n),简化了对两体函数最佳参数的搜索;③幂函数与衰减截断函数的组合可以生成弹性峰形状和不同的径向分布。
1.4 SSW-NN
密度泛函理论与势能面采样方法(如遗传算法、盆地跳跃、随机表面行走SSW 等)的结合已成为解析结构和搜索反应活性位点的有力表征工具。与其他计算方法(如力场、耦合簇)相比,它们在催化方面的成功是由于它们在可接受的计算时间内可以提供可靠的计算结果。可接受的计算时间主要是由于使用了便宜的泛函(如PBE、PW91)和较小的计算模型(<100个原子)。目前,大多数势能面搜索的DFT 计算都是在小模型上进行的,这些小模型足以描述简单的催化系统。对于费托(F-T)催化剂上的长碳链生长过程(C>5)、分子自组装、无定形结构搜索等复杂体系来讲,由于计算时间的限制,DFT无法处理这样复杂的体系。神经网络的高效和准确性可大大弥补DFT 的不足,NN 比DFT 至少快3~4 个数量级,同时保持能量和力的精度与DFT 相当。用神经网路势函数结合势能面搜索方法来对势能面结构进行搜索是解决复杂催化体系的强有力的工具。本文作者课题组开发的SSW 和NN(SSW-NN)相结合可以实现对势能面的快速搜索。完整的SSW-NN 方法的完整使用过程可分为四个步骤:产生DFT数据集、NN 势能面拟合、SSW-NN势能面搜索和DFT验算。
(1)产生DFT 数据集 本步骤的意义在于获取足够多的数据集,方便后续NN 训练使用。在此,根据经验教训提出三点注意事项:①训练集一定要多样化,包括不同的原子数、不同的化学计量比、不同的原子配位环境(体相、表面、团簇)等;②训练集的DFT 计算参数一定要采用高精度计算,包括平面波能量截断、k 点网格密度等;③不同训练集保证相同的计算参数,方便某些特殊需求时可以便捷地合并训练集。
(2)NN 势能面拟合 本步骤的意义在于利用NN 拟合DFT 训练集得到NN 势函数。NN 拟合原理及注意事项见上文。表1 列出了常用的NN 拟合过程中的一些超参数。

表1 NN拟合常用超参数
(3)SSW-NN势能面搜索 本步骤的意义在于利用NN势函数结合SSW快速搜索势能面。需要注意的是在进行SSW-NN 之前,一定要对NN 势函数进行精度分析,确保所得结果准确可靠。此外,由于NN势函数与DFT所得结果之间存在误差,可能会导致部分结构能量顺序翻转,这就要求SSW-NN搜索完得到的结果必须要进行DFT验算。
(4)DFT 验算 本步骤的意义在于检验SSWNN 模拟得到的结果的可靠性。验算时DFT 的参数设置要求与步骤(1)的训练集参数设置一样。另外,验算时不能只检查所关心的结构(通常是势能面极小值),还需验算与其能量相近的结构,确保没有发生势能面极小点预测错误的现象。
此外,为了获得一个稳健可靠、高精度、容错率高的NN势函数,在训练过程中可以通过自学习过程来实现,见图4。步骤就是上述(1)~(4)过程来回迭代。每得到一次NN 势函数就进行SSW-NN 搜索,将一些未能学好的结构重新进行DFT 计算添加到原始数据集中,然后再训练一次产生新的NN势函数。直到最后产生的NN 势函数进行SSW-NN模拟时不会有错误结果出现。
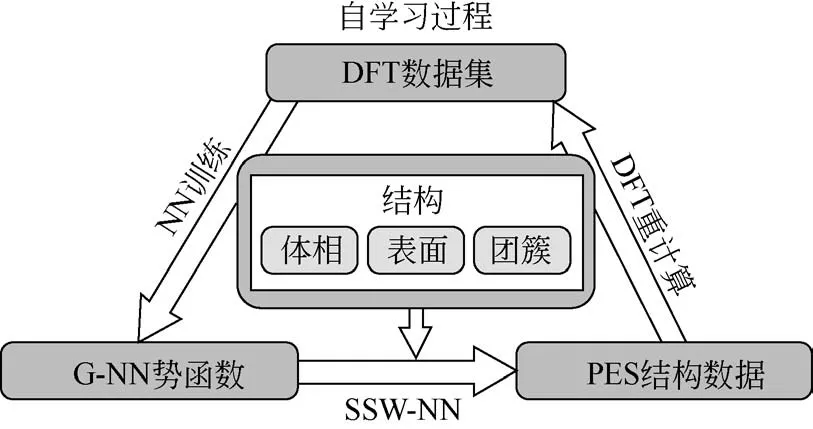
图4 NN势函数自学习过程
2 SSW-NN方法应用
2.1 二氧化钛全局势能面搜索及结构预测
二氧化钛(TiO2)半导体材料以其成本低、催化活性高、化学稳定性高、抗氧化能力强、安全无毒以及无二次污染等优点被公认为是最具有开发前景的环保型材料之一[35-37]。迄今为止,研究人员已经发现了十几种不同的二氧化钛晶相,从常见的三维晶相(金红石rutile、锐钛矿anatase、板钛矿brookite 等)到二维晶相(纤铁矿型lepidocrocite-TiO2)。其中,比较独特的是斜方锰矿型ramsdellite-TiO2(TiO2-R)[38]和锰钡矿型hollandite-TiO2(TiO2-H)[39]晶相含有一维孔道结构,孔径在3~5Å(1Å=0.1nm)。由于Ti4+的可还原性和其便于离子迁移的孔道结构,使得它们已经被开发为锂(钠)离子电池的负极材料[40-42]。当然,比TiO2-R和TiO2-H具有更大孔径的材料也被开发出来了,比如,多孔TiO2膜(平均孔径约9Å)[43-44]、TiO2干凝胶(平均孔径15Å)[45],但这些孔要么是粒间孔要么是无定形孔,使得这些微孔很容易在高温下(400oC)坍塌掉。因此,是否存在稳定的、具有更大孔径的二氧化钛新晶相仍旧是一个未知之谜。通过对TiO2势能面进行全局搜索可以回答上述疑问。
采用SSW 方法,从TiO2的不同晶体结构出发[金红石、锐钛矿、板钛矿、TiO2(B)等],搜索其周围可能存在的晶相,得到103个势能面极小点。基于这些势能面极小点,绘制出了二维TiO2势能面E-OP 图,见图5。其中E 为相对与锐钛矿的能量,OP 为Steinhardt 类型的有序结构参数(order parameter)。OP可以很好地区分势能面的结构,具体计算见式(15)。

从图5(a)中可以看出,整个二氧化钛的势能面底部可以看成一个大的“W”。其中常见的金红石、锐钛矿、TiO2(B)等均处于“W”底的左半部分(OP=0.3~0.6)。这一部分的典型特征就是所有晶相均是由[TiO6]八面体组成的。而很有意思的是,“W”底的右半部分(OP=0.6~0.9)却从来没有人报道过。这右半部分结构的典型特征就是其组成单元为[TiO5]三角双锥体(trigonal bipyramid,TB)。在这些结构中发现了一系列的微孔TiO2晶体。这些晶体均由[TiO5]三角双锥体组成。通过不同的组合方式可以形成六元环和八元环微孔,孔径5.6~6.7Å。传统上一直认为特殊的[TiO5]配位构型是热力学不稳定的,但DFT-PBE计算结果表明这些TiO2(TB)晶体竟然比常见的金红石相还要稳定,而且从从头算分子动力学模拟中也看出这些晶体在高温下是很稳定的。它们稳定的最关键原因是由于Ti-O 晶格中形成的强离子键作用,补偿了[TiO5]中弱的共价键相互作用。通过对锂离子嵌入能量和结构参数进行评估,预测这些微孔二氧化钛晶体是良好的锂离子负极候选材料。
2.2 无定形TiOxHy势能面搜索及结构预测
二氧化钛(TiO2)是一种很有前途的光催化剂。然而,受限于宽的带隙(>3eV),使得其吸光效率大大降低,只能吸收紫外光。因此,众多的研究人员试图通过各种办法来减小二氧化钛的带隙提高催化活性。2011 年,Chen 等[46]取得了重大突破,他们获得了一种黑色TiO2材料。“黑色”就意味着这个材料在整个可见光区都有很强的吸收能力,并且这个材料表现出很高的催化活性。据报道,黑色TiO2的析氢反应(HER)活性要比普通半导体光催化剂(如商业用P25)的活性高出好几个数量级[47]。更重要的是,合成黑色TiO2的方法也非常简单,用含氢的还原剂(如H2和NaBH4)处理原始TiO2材料即可[48-49]。因此,基于Chen 等的结果,后续有大量的研究改变合成条件合成各种各样的黑色TiO2或者理解为什么黑色TiO2材料可以如此明显地提高HER 活性。然而,令人遗憾的是,经过这么多年的探索,对机理的理解知之甚少。其中的障碍在于结构的复杂性。大量研究表明,黑色TiO2在加氢过程中涉及复杂的结构演化,伴随着一系列从TiO2到TiOxHy的相变,导致表面无定形化。在化学和材料科学中,探索无定形表面的结构本身就是一项极具挑战性的任务,更别提确定催化活性位点。为了解决HER 在无定形表面的活性位点,不仅需要精确的TiOxHy势能面(PES),而且需要一个强有力的采样工具能够遍历各个可能的结构。因此利用SSW-NN 方法可以实现探索二氧化钛加氢后无定形壳层的结构,理解结构无定形化机理,从而确定HER 的活性位点。利用SSW-NN,探索TiO2Hx和TiOx的相空间(含有11~16 个原子),氢化的TiO2(112)表面相空间(Ti56O112Hx)。每个配比搜索得到了最少1万个局域最小值结构,其中包括从晶体结构到非晶结构,定量确定了不同温度和H2压力下的TiOxHy结构与组成的热力学相图。

图5 48原子的TiO2全局势能面等值线图及TiO2晶体结构
在各种锐钛矿TiO2表面上,只有在(112)表面上加氢会逐渐导致结构无定形化。如图6(a)所示,第一层中近乎一半的Ti 原子改变了它们的位置。特别地,25%的Ti5c原子向上隆起,变成了Ti4c原子,另外25%的Ti5c原子向下沉陷到第二层,变成Ti6c原子。在最终重构完的表面上,Ti—O 键的长度分布明显宽化,从1.80Å到2.2Å,而在未重构的(112)表面为1.9~2.1Å。(112)表面完全失去原来的锐钛矿的键合模式,重构成了无定形结构。进一步分析表面H 浓度发现,在H 覆盖度较低[≤0.19ML(单分子层)]时,(112)表面仍旧可以维持规整的表面结构,所有添加的H原子都位于表面O2c上。H 浓度高于0.19ML 时,表面开始重构,整个重构过程是热力学有利的。最大放热发生在表面覆盖0.69ML 的H 原子,这与未重构表面形成了鲜明对比。未重构TiO2-0.69H 表面的能量要比重构完的TiO2-0.69H 表面能量高3.64eV/(4×2)表面。这种高氢覆盖的表面不仅使无定形TiO2呈现黑色,而且为HER 反应提供了前所未有的低能垒反应通道:暴露的Ti 原子上可以形成瞬时Ti-H 氢化物。在未重构表面,两个OH耦合形成H2。由于两个相邻的O2cH 之间的距离比较长(初始状态下的H-H 距离为3.6Å),导致反应具有非常高的势垒2.8eV。而在无定形TiO2-0.69H 表面,存在一条全新的低能垒的反应通道,通过一种全新的TiH/OH耦合机制产H2。与传统的OH/OH 耦合通道相比(能垒>1.6eV),TiH/OH耦合反应具有更低的势垒,即0.6eV[图6(b)]。电子结构分析表明TiH 氢化物的形成使得电子从高能的Ti 3d 轨道转移到H 原子上形成H-离子,而H-离子可以很容易地与吸附在O上的H+离子结合形成H2。H-NMR 谱证明在氢化TiO2表面TiH 基团的形成[50]。研究结果不仅对无定形材料特有的表面形态和反应提供了深入的了解,而且证明了利用NN势函数的全局采样方法对于解决实际反应下的复杂结构具有广阔的应用前景。
2.3 ZnCrO 催化剂上的合成气转化制甲醇活性位点探索

图6 无定形TiO2-0.69H表面结构及(112)和无定形表面的H耦合产H2能量曲线
锌-铬氧化物(ZnCrO)可被用来催化合成气转化制甲醇或其他化学物质[51-52],并可作为合成气转化制烯烃的氧化物-沸石复合催化剂的关键成分[53]。作为第一代合成气制甲醇的工业催化剂[54],自20 世纪30 年代以来,人们便对ZnCrO 催化剂进行了广泛的研究。目前普遍认为高温煅烧后形成的最稳定晶相为尖晶石晶型的ZnCr2O4[55]。许多研究小组已经表明,Zn∶Cr比对合成气制甲醇的催化活性和选择性有显著影响[51,56-59]。当Zn∶Cr比为1∶1时,催化剂通常能达到最佳的活性和选择性,例如,甲醇的收率约为90g/(kgcat·h),选择性为80%[59]。而纯ZnCr2O4尖晶石催化剂的活性和选择性相对较差,甲醇产率小于5g/(kgcat·h)且甲醇选择性为14%(烷烃的选择性约为45%)。然而,Zn∶Cr>1∶2 时ZnCrO结构不确定,目前还没有关于ZnCrO精确结构的数据。对活性中心的了解不足,阻碍了合成气在ZnCrO催化剂及相关氧化物分子筛催化体系上催化性能的进一步优化。为了将ZnCrO的原子结构与其催化活性联系起来,必须探索三元体系的结构空间,并确定与反应条件有关的结构。因此,可以利用SSW-NN 方法来探索ZnxCryOz的结构,从而确定其体相和表面结构。基于SSW-NN方法,每个Zn∶Cr∶O 配比搜索得到最少1 万个局域最小值结构,其中包括从晶体结构到非晶结构[60]。

图7 ZnCrO势能面结构的热力学结果及其表面的合成气转化路线
首先,对体相ZnCrO 结构的热力学相空间进行扫描,其中Zn∶Cr∶O 比从CrOx延伸到ZnO,见图7(a)的Zn-Cr-O 三元相图。尖晶石型晶体结构为ZnCrO 的主要结构框架,范围从Zn∶Cr=0∶1到Zn∶Cr=1∶1。实验上已知的ZnCr2O4晶体属于该区域。这些晶体具有由O2-阴离子形成的面心立方(fcc)子晶格,在间隙四面体(Td)和八面体(Oh)位点具有不同的Zn 和Cr 占据。进一步计算了不同Zn∶Cr比的ZnCrO化合物的形成能并绘制得到热力学凸图,见图7(b)。只有少量的具有尖晶石型骨架的ZnCrO 成分,即Zn3-xCr3+xO8(0≤x≤1)和ZnxCr4O8(1.5≤x<2),具有负的形成能,表明它们在实验上有可能被合成出来。体相热力学结果指出这一范围内的两个关键组分,即Zn∶Cr=1∶2(原子数比)和1∶1。其中,Zn3Cr3O8晶相中[ZnO6]0h的浓度最高,是Zn∶Cr>1∶2 化合物的最佳代表。ZnCr2O4中的[CrO6]Oh浓度最高且只有[ZnO4]Td,不含[ZnO6]0h。
进一步的表面结构分析表明,Zn3Cr3O8和ZnCr2O4的最稳定晶面分别是(0001)和(111)晶面。虽然这两个表面的标记不一样,但它们显示出相同的第一层表面结构,即由Zn-O-Cr 相互连接形成的蜂窝状六元环。其中暴露了三配位的Zn3c、三配位O3c和六配位Cr6c原子[图7(c)]。而这两个表面最主要的区别在于第二层的Zn配位。在Zn3Cr3O8中Zn 离子占据了Td和Oh位置,但在ZnCr2O4中Zn离子仅占据Td位置。由于不同的次表层结构导致这两个表面在反应气氛下的氧空位浓度不同,Zn3Cr3O8表面不仅在表面可以产生0.25~0.5ML(单分子层)浓度的Ov,而且还可以产生0.25ML 的次表层Ov。而ZnCr2O4的表面Ov浓度在O0v.25ML~O0v.5ML之间。特殊的次表层氧空位导致表面暴露的活性位点完全不同,Zn3Cr3O8表面会暴露两个采用平面构型的四配位Cr4c原子,而ZnCr2O4表面会暴露出两个五配位Cr5c原子。
基于以上确定好的表面活性位点,进一步评估ZnCr2O4和Zn3Cr3O8两种催化剂上的合成气转化活性[图7(d)]。合成气转化路径为:CO—>CHO—>CH2O—>CH3O—>CH4/CH3OH,反应的决速步为CH3O 加氢。在Zn3Cr3O8表面,产生CH3OH 在动力学上更容易一些,能垒仅为1.33eV。而在Zn3Cr3O8表面,CH3O 加氢更倾向于形成甲烷(反应能垒1.75eV),而非甲醇。产生这种活性差别的本质原因是由于不同的CH3O 吸附:在Zn3Cr3O8表面,弱的CH3O 吸附导致CH3OH 的生成,而ZnCr2O4表面,强的CH3O 吸附则产生CH4。基于以上对催化活性中心的分析,可以很好地解释为什么Zn:Cr 比变化会极大地影响催化活性和选择性。
3 结语
本文概述了神经网络全局势函数方法及其在多相催化中的应用。从确定反应活性位点到理解反应机理,神经网络在多相催化领域能够大放异彩,完全有赖于合理的、多样和完备的DFT 数据集。未来拓展神经网络势函数计算的工作重点之一就是获取更多的有效数据集,建立数据库。此外,神经网络目前主要集中在固体材料的结构搜索,下一步将逐渐构建化学反应势能面。LASP 软件中目前提供的反应势函数覆盖了Ⅷ过渡金属和简单的小分子反应,比如PtCHO(见www.lasphub.com)。考虑到表面反应的复杂性,在此提出几个未来可能的研究方向。
(1)神经网络拟合化学反应 该领域面临的问题是如何获得足够多的化学反应过渡态区域的数据集。通常的采样方法主要集中在势能面底部,而对于过渡态区域的采样很少。因此需要有特殊的方法来增强过渡态区域的采样。
(2)多元素体系的神经网络拟合 目前HDNN的架构是每种元素有着各自的神经网络,随着元素种类的增多,神经网络的参数会变得很多,训练及使用过程会变得缓慢,数据集构造也会变得更为复杂,因此需要开发新型的神经网络架构来满足多元素体系的使用。
(3)神经网络拟合其他性质 目前神经网络主要是获得结构与能量之间的关系,今后可以拟合其他电子结构数据,如电荷、带隙等。
猜你喜欢
——《势能》
文化纵横(2022年3期)2022-09-07
中学生数理化·八年级物理人教版(2022年6期)2022-06-05
现代电力(2022年2期)2022-05-23
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
中学生数理化·八年级物理人教版(2021年6期)2021-11-22
电子制作(2019年19期)2019-11-23
中学生数理化·八年级物理人教版(2019年6期)2019-06-25
电子制作(2019年24期)2019-02-23
