
掺杂对新型二维材料磷烯光电性质的影响
2021-02-25 06:03张忠政张春红闫万珺覃信茂
量子电子学报 2021年1期
张忠政, 张春红, 闫万珺, 覃信茂
(1 安顺学院数理学院, 贵州 安顺 561000;2 安顺学院电子与信息工程学院, 贵州 安顺 561000;3 安顺学院航空电子电气与信息网络工程中心, 贵州 安顺 561000)
0 引 言
自石墨烯问世以来,二维材料逐渐成为研究热点。然而由于石墨烯没有本征半导体带隙,限制了其在半导体材料领域的应用。继石墨烯、二硫化钼之后,张远波课题组发现了具有独特褶皱结构的磷烯材料[1]。众多学者关注并研究了二维材料磷烯的结构和优点。研究发现:磷烯是直接带隙半导体材料,不仅具有良好的电导率和热导率以及各向异性等优点,而且具有高载流子迁移率[2-8]。这些性质使得磷烯材料在光电材料领域备受关注并显示出巨大的应用潜力。
吸附和掺杂是调制材料性能的主要技术。如Cai 等[9]采用第一性原理方法对小分子CO、H2、H2O、NH3、NO、NO2和O2吸附磷烯的能量及电荷转移和磁性进行了研究。Hu 等[10]对Li、Na、Mg、Al、Cr、Fe、Co、Ni、Mo、Pd、Pt 和Au 吸附磷烯的结构、吸附能、生长模式、扩散势垒、磁性、偶极矩和功函数进行了研究。Jing 等[11]研究了三种典型有机分子吸附单层磷烯的电子和光学性质,发现磷烯的带隙会由于分子修饰而显著变窄,外加电场可对带隙进行进一步调制;表面分子修饰是增强磷烯在各个方向集光的有效方法。He 等[12]研究了亲电分子吸附单层磷烯的电荷转移行为。Srivastava 等[13]研究了空位和C、N、O、Fe、Co、Ni 吸附磷烯的电磁性质,发现了吸附后的磷烯材料能够对费米能级进行控制从而实现对磁矩的调节。Huang 等[14]研究了在不同浓度掺杂情况下N、As、Sb、Bi 掺杂对磷烯电子性质的影响。Khan 等[15]对Al、Si、S、Cl 四种非磁性原子掺杂磷烯进行了研究,发现Si、S、Cl 三种原子掺杂后,磷烯具有金属特性且有磁性,而Al 掺杂后磷烯仍属半导体且无磁性。Seixas 等[16]研究得出当磷烯掺杂Co 的含量达到2.7%时,磷烯的磁学性质发生变化,材料的居里温度变为466 K。
综上可见,对于磷烯材料改性的研究主要在电学和磁学性质方面,而在光学性质方面的研究却很少。研究发现通过掺杂C 和Al 能够改变原有材料的光电性质[17-22]。本文采用第一性原理赝势平面波方法,对新型二维材料磷烯进行了掺杂改性研究。所选择掺杂元素是C 和Al,对两种元素掺杂后的磷烯在物理结构和光电性能方面进行了全面的计算与机理分析。
1 计算模型和方法
黑磷晶体的晶格常数为a= 0.4376 nm,b= 1.0478 nm,c= 0.3314 nm[23]。磷烯是从晶体中剥离出来的单层黑磷。计算用的磷烯模型是含有16 个P 原子的超胞,如图1(a)所示。杂质掺杂磷烯的模型是用一个杂质原子X(X=C,Al)置换图1(a)中坐标为(0.29,0.40,0.50)的一个P 原子后得到的,如图1(b)、(c)所示。
采用第一性原理赝势平面波方法进行计算。用USPP[24]来处理离子实与电子间的相互作用,采用GGA 的PBE 泛函[25]来处理电子间的交换关联能。设置Ecut-off= 310 eV,精度为2×10-6eV/atom,参与计算的价电子选取如下:C 的2s22p2,Al 的3s23p1,P 的3s23p3。Brillouin zone 积分采用4×4×4 的Monkhorst-Pack 形式[26]的对称特殊k点方法,设置40×45×30 的FFT 网格参数。
结合能Eb的表达式为[27]
式中ET为掺杂体系的总能量,np为体系中P 原子的个数,E(P)为单个孤立P 原子的总能量,E(X)为单个孤立掺杂原子X的总能量,n为体系中总的原子个数。
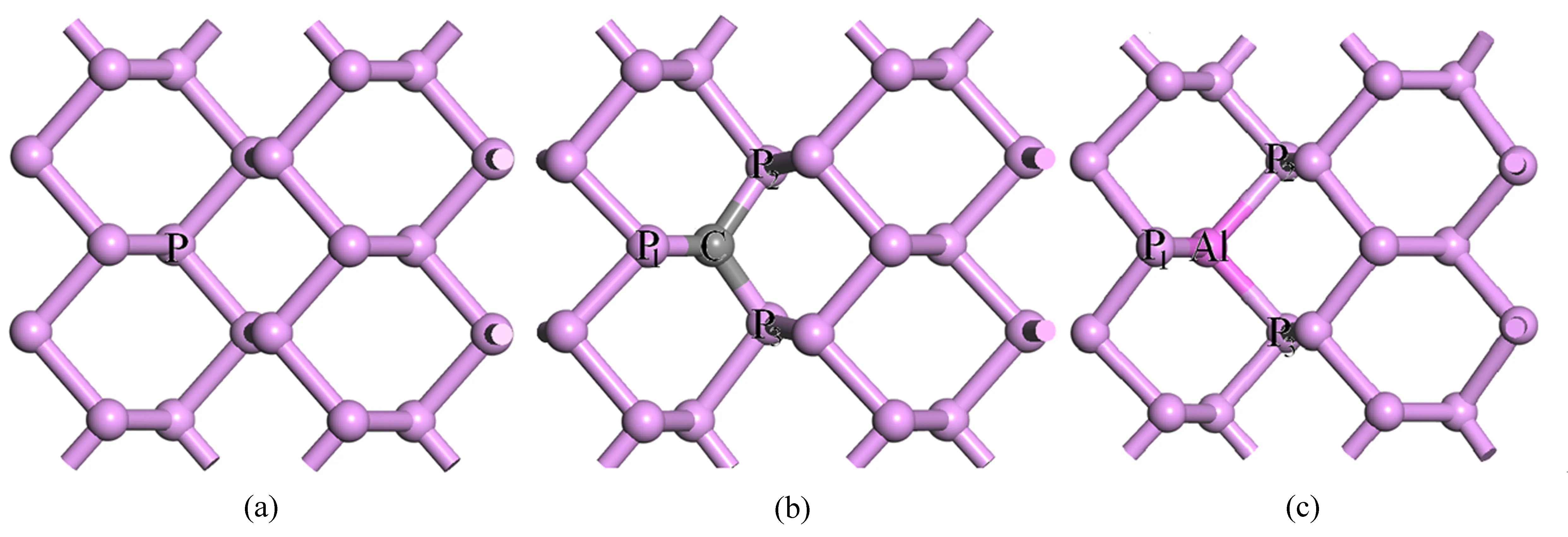
图1 新型二维材料磷烯模型。(a)未掺杂;(b)碳掺杂;(c)铝掺杂Fig.1 Model of new two-dimensional material phosphorene.(a)Undoped;(b)C doped;(c)Al doped
2 计算结果与讨论
2.1 几何结构
表1 列出了杂质X掺杂新型二维材料磷烯的结构参数和结合能。其中dX-Pi(i=1,2,3)代表X原子与三个相邻P原子的键长(如图1 所示),Eb为体系的结合能。
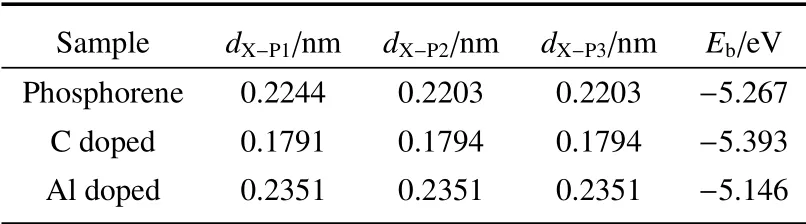
表1 新型二维材料磷烯的结构参数和结合能(X=C,Al)Table 1 Structural parameters and binding energy of new two-dimensional material phosphoene(X=C,Al)
由表1 可以看出:计算采用的磷烯模型在同一平面上的P-P 键长均相等,为0.2203 nm,与实验值[28]0.2224 nm 的误差不超过0.27%,而不在同一平面上的P-P 键长的计算值为0.2244 nm,与实验值[28]0.2244 nm 一致,说明计算采用的磷烯模型是可靠的。
由图1 可以看出:C 和Al 掺杂后,新型二维材料磷烯仍然具有层状褶皱结构,但是掺杂后磷烯的结构发生了畸变。这些变化可以由表1 中给出的dX-P1、dX-P2、dX-P3三个键长发生了变化而直接看出。造成键长发生变化的原因是各原子的半径不同,如C、Al、P 的原子半径分别为0.086、0.143、0.130 nm。由于C 原子的半径比P 原子的小,所以C 掺杂后与相邻P 原子的键长dX-Pi变短;而Al 原子的半径比P原子的大,所以Al 掺杂键长dX-Pi变长。同时,C 掺杂后引起的磷烯结构畸变比Al 掺杂后要严重,这是因为C 和P 的原子半径差比Al 和P 的原子半径差要大得多。表1 中列出的与杂质(X=C,Al)相连的3 个P 原子之间的键长dX-Pi与文献[29-31]的研究结果符合得很好,说明基于此掺杂结构计算出来的光电性能的结果是可靠的。
由表1 中列出的掺杂前后磷烯体系的结合能数值来看,C 和Al 掺杂后,体系的结合能Eb均为负值,且与未掺杂时体系的结合能数值接近,说明掺杂体系的结构是稳定的。
2.2 能带结构
图2 为新型二维材料磷烯掺杂前后的能带结构图。由图2(a)可以看出:磷烯是具有0.909 eV 的直接带隙半导体,价带顶和导带底都在布里渊区的Γ 点处,这与Seifert 等[32]的计算值0.9 eV 非常吻合,说明计算方法及精度是可靠的。C、Al 掺杂后,对磷烯的能带结构带来了很大的影响。由图2(b)可以看出,C掺杂后费米能级进入价带中,在Γ 点处的价带顶上移,导带底的位置基本不变,使得掺杂后体系的带隙变窄,变为0.826 eV 的直接带隙。由图2(c)可以看出,Al 掺杂后体系变为间接带隙半导体,带隙略有展宽,带隙为0.965 eV 且在Γ 点以外取得。
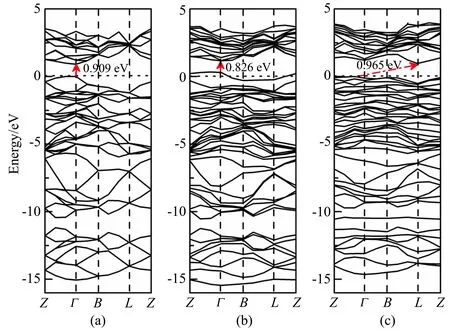
图2 磷烯的能带结构。(a)未掺杂;(b)碳掺杂;(c)铝掺杂Fig.2 Band structure of phosphorene.(a)Undoped;(b)C doped;(c)Al doped
2.3 Mulliken 布居分析和差分电荷密度
与杂质原子X相邻磷原子的Mulliken 布居分析列于表2 中。
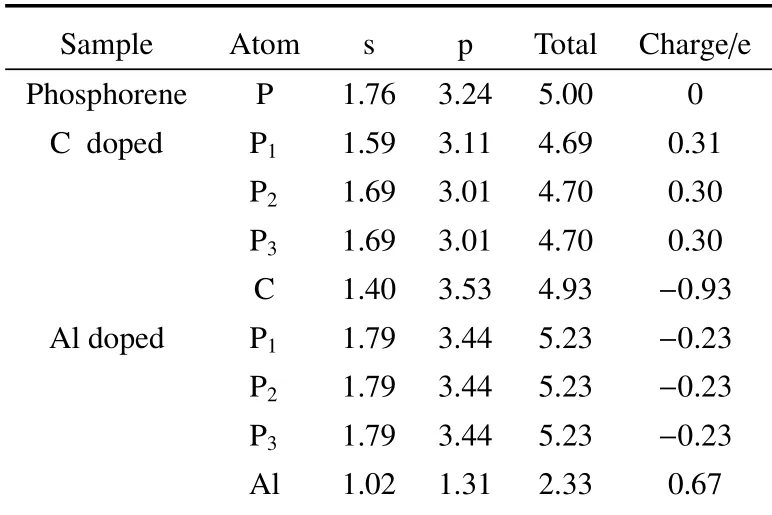
表2 与杂质原子相邻磷原子的Mulliken 布居分析Table 2 Mulliken population analysis of P atom around the impurity atom
由表2 可知,磷烯体系的P 原子之间是不发生电荷转移的,P-P 键是典型的共价键。但是当杂质X掺入后,如与C 原子相邻的3 个P 原子电荷减少了(0.30 e 或0.31 e),这些电荷正是C 原子从相邻的3 个P原子上得到的(0.93 e),说明C 掺杂后体系的电荷分布发生了转移,电荷从P 原子转移到C 原子。与C 掺杂情况相反的是,Al 掺杂后,如与Al 原子相邻的3 个P 原子电荷都增加了(0.23 e),这些电荷正是Al 原子失去的电荷(0.67 e),说明Al 掺杂后体系的电荷是从Al 原子转移到相邻的P 原子上。以上分析的电荷分布转移情况也可由如图3 所示差分电荷密度图直接看出。
图3 为杂质X掺杂前后新型二维材料磷烯的差分电荷密度图,其中红色区域表示电荷积累的空间分布,蓝色区域表示电荷消耗的空间分布。由图3 可以直接看出,杂质周围的电荷分布发生了变化。C 掺杂后,C 原子附近出现了电荷积累,而与C 相邻的3 个P 原子附近出现了电荷的消耗;Al 掺杂后,Al 原子附近出现了电荷消耗,而与Al 相邻的3 个P 原子附近出现了电荷的积累。这些差分电荷分布的结果与上面的布居分析结果一致。
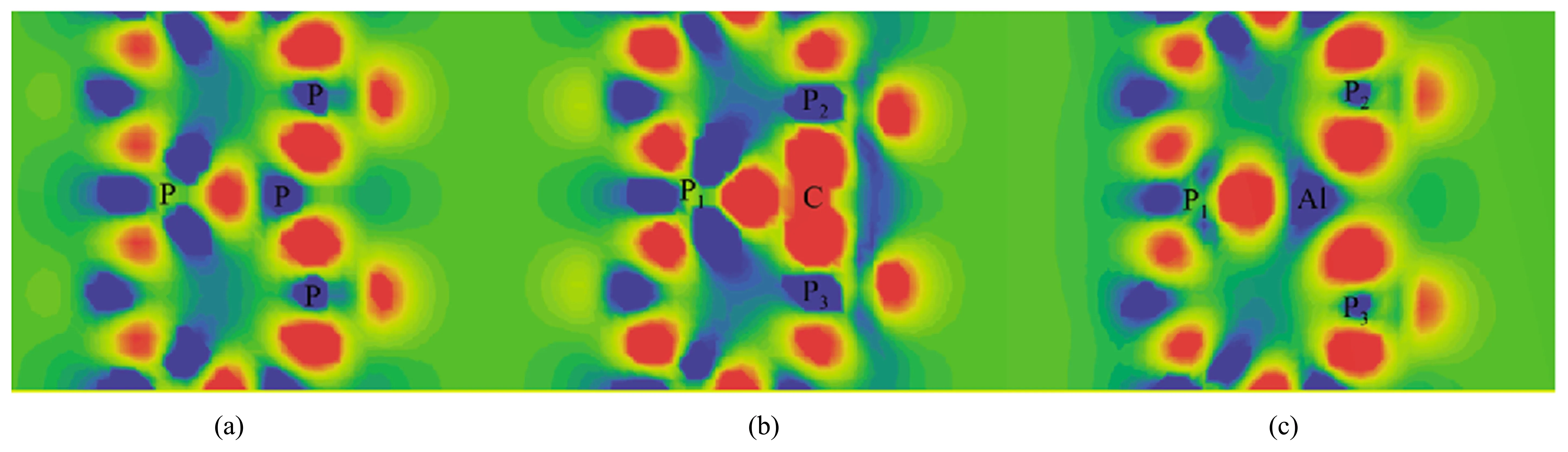
图3 磷烯的差分电荷密度图。(a)未掺杂;(b)碳掺杂;(c)铝掺杂Fig.3 Electron density difference of phosphorene.(a)undoped;(b)C doped;(c)Al doped
2.4 光学性质
为探索杂质X掺杂对新型二维材料磷烯光学性质的影响,进一步计算了掺杂情况下在(1 0 0)极化方向上磷烯的复介电函数、吸收系数、反射率、折射率及能量损失函数等光学性质。
图4 展示了杂质X掺杂前后新型二维材料磷烯的复介电函数。由图4(a)可以看出:新型二维材料磷烯的静态介电常数ε1(0) = 6.02。C 掺杂后,静态介电常数减小为5.11;Al 掺杂后,静态介电常数增大为6.87。还可以看出:在E<1.94 eV(红光及红外线)的范围内,C 掺杂后介电函数的实部ε1(ω)明显减小,而Al 掺杂后明显增大,说明C 掺杂后减弱了磷烯储存电磁能的能力,而Al 掺杂后增强了磷烯储存电磁能的能力。由图4(b)可以看出:C、Al 掺杂后,磷烯材料在E= 4.79 eV 的介电函数虚部的主峰ε2均降低,这个介电峰主要是由P 的3p态电子和少量C 的2p态电子以及Al 的3p态电子贡献的。该介电峰降低是因为C 的2p态电子和Al 的3p态电子均与p的3p态电子发生了轨道杂化。以上结果说明采用不同的杂质掺杂可以调制磷烯材料的介电性能。
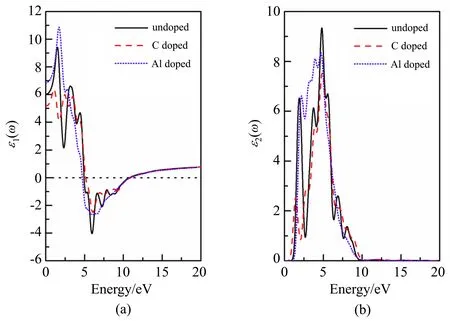
图4 磷烯的复介电函数。(a)ε1(ω);(b)ε2(ω)Fig.4 Complex dielectric functions of phosphorene.(a)ε1(ω);(b)ε2(ω)
图5 是杂质X掺杂前后新型二维材料磷烯的光学性质谱。图5(a)是吸收系数,可以看出掺杂对磷烯材料光学吸收端的影响与前面能带结构图给出的带隙分析是一致的。而且掺杂后主吸收峰与介电函数虚部的主峰降低的原因相同。图5(b)是反射系数,可以看出,在(1 0 0)极化方向上磷烯材料的反射率峰值非常高,材料表现出金属的特性。当C、Al 掺杂后磷烯材料的反射率峰值明显降低。图5(c)是折射率,由图可以看出,磷烯的静态折射率n0=2.46,C 掺杂后n0减小为2.26,Al 掺杂后n0增大为2.62。对于由磷烯材料制成的光学器件,当对其折射率有一定数值要求时,这个结果可供参考。图5(d)是损失函数,虽然C 掺杂后材料的损失函数峰值略有增加,Al 掺杂后材料的损失函数峰值明显降低,但是掺杂前后磷烯材料对光的损失都局限于10.80 eV 附近能量范围内,说明掺杂前后磷烯材料都可作为光储存材料。
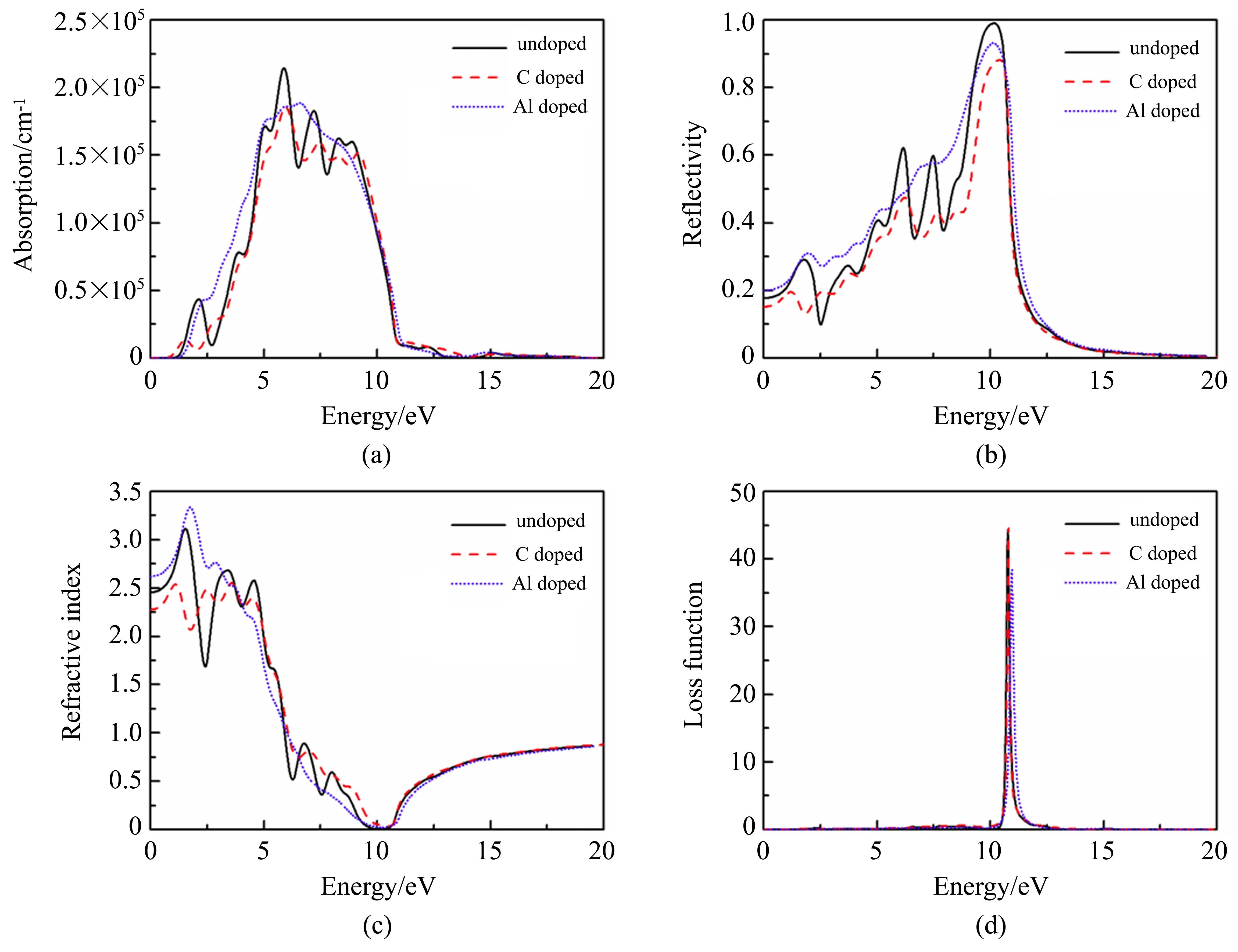
图5 磷烯的光学性质谱。(a)吸收系数;(b)反射率;(c)折射率;(d)能量损失函数Fig.5 Optical properties of phosphorene.(a)Absorption;(b)Reflectivity;(c)Refractive index;(d)Loss function
3 结 论
利用第一性原理赝势平面波方法计算了杂质(X=C,Al)掺杂新型二维材料磷烯的结构参数、能带结构、Mulliken 布居分析、差分电荷密度以及光学性质。结果表明:杂质掺杂后磷烯材料的结构发生了畸变,但是掺杂体系的结构是稳定的。C、Al 掺杂后,对磷烯的能带结构带来了很大的影响。C 掺杂后,费米能级进入价带中,在Γ 点处的价带顶上移,导带底的位置基本不变,使得掺杂后体系的带隙变窄,变为0.826 eV 的直接带隙;Al 掺杂后,体系变为间接带隙半导体,带隙略有展宽,带隙为0.965 eV 且在Γ 点以外取得。Mulliken 布居分析和差分电荷密度的分析都表明掺杂后体系的电荷分布发生了转移,C 原子附近出现了电荷积累,而Al 原子附近出现了电荷消耗。在(1 0 0)极化方向上的光学性质计算表明:在红光及红外线的范围内,C 掺杂后磷烯材料储存电磁能的能力有所减弱,而Al 掺杂后储存电磁能的能力有所增强;C 掺杂后折射率n0减小,Al 掺杂后折射率n0增大;吸收系数和反射率峰值均降低;掺杂前后磷烯材料对光的损失都局限于10.80 eV 附近的能量范围内,说明掺杂前后磷烯材料都可作为光储存材料。以上结果说明采用不同的杂质掺杂可以调制磷烯材料的光电性质。
猜你喜欢
原子与分子物理学报(2022年3期)2022-03-05
辽宁科技大学学报(2021年2期)2021-07-22
艺术品鉴(2020年6期)2020-12-06
中学生数理化(高中版.高考理化)(2020年10期)2020-10-27
青岛大学学报(工程技术版)(2019年2期)2019-09-10
领导文萃(2017年6期)2017-03-24
新高考·高一物理(2016年7期)2017-01-23
中学生数理化·高一版(2016年7期)2016-12-07
中学化学(2015年8期)2015-12-29
新高考·高一物理(2015年6期)2015-09-28
