
HMGB1-TLR4/RAGE信号通路在ALI/ARDS中的研究进展
2021-06-08 07:00秘乐范绍辉徐宇舒小燚王红嫚
国际呼吸杂志 2021年9期
秘乐 范绍辉 徐宇 舒小燚 王红嫚
遵义医科大学第五附属(珠海)医院呼吸与危重症医学科519100
急性肺损伤 (acute lung iniury,ALI)/ARDS是非心源性因素导致的肺泡上皮细胞及毛细血管内皮细胞损伤,导致急性、顽固性呼吸窘迫。2012年柏林定义取消了ALI命名,将本病统称为ARDS[1],但在动物实验研究中暂无统一的命名标准。其发病机制复杂,尚无有效治疗措施。高迁移率族蛋白B1 (high-mobility group box 1 protein,HMGB1)可能是启动、维持和加剧瀑布式炎症级联反应的重要晚期炎性因子[2-4]。当细胞外的HMGB1结合到细胞表面受体时,如晚期糖基化终产物受体 (receptor for advanced glycation end products,RAGE),Toll 样受体(Toll-like receptors,TLR)家族中的TLR2、TLR4[5]、TLR9[6]及趋化因子CXCL12[7]等,可激活下游通路,诱导转录多种炎性因子[8]。近年来,HMGB1-TLR4/RAGE信号通路在ALI/ARDS中的作用逐渐引起大家关注,而寻找炎症相关的潜在分子机制可能有助于确定ALI/ARDS新的治疗靶点 (图1)。
1 HMGB1与ALI/ARDS
1.1 HMGB1的结构、分布与功能 人类HMGB1基因位于13q12染色体上。HMGB1 是一种有215 个氨基酸的单链蛋白,由A-box、B-box和C-tail 3个结构域组成,其中B-box具有炎症活性,而A-box对B-box有一定的拮抗作用[9]。HMGB1 广泛存在于淋巴、脑、肺、肝、心、肾、脾等组织细胞中[10]。正常生理条件下,HMGB1作为一种胞核内DNA 结合蛋白,表达量稳定在基础水平,在基因转录、基因调节及DNA 修复重组等过程中起重要作用[9,11]。在细胞应激、活化或凋亡、死亡过程中,HMGB1转移至胞质和胞外基质[12-13],与相应受体结合,介导TLR4/RAGE等众多下游信号通路参与免疫炎症反应。
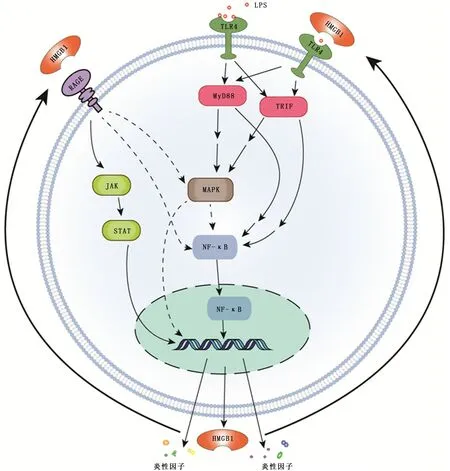
图1 HMGB1-TLR4/RAGE相关信号通路
1.2 HMGB1在ALI/ARDS中的作用
1.2.1 HMGB1 放大炎症级联反应 HMGB1 作为ALI/ARDS炎症反应进程中重要的晚期炎性因子,从胞核移至胞质,再由炎症细胞将其释放到胞外,可诱导炎性因子释放、细胞聚集[13]、细胞分化[14]、细胞极化[5]等多种反应。胞外HMGB1 与细胞表面受体结合,如RAGE、TLR2、TLR4[5]、TLR9[6]等,进一步激活下游通路而发挥其生物学效应。此外,HMGB1可调节单核细胞的迁移[13],介导内皮细胞的高通透性[15],并激活巨噬细胞[13]、树突状细胞[16]、中性粒细胞等[5],使这些细胞释放单核细胞趋化蛋白1 (monocyte chemotactic protein 1,MCP-1)、肿瘤坏死因子α (tumor necrosis factor-α,TNF-α)、IL-1β、IL-6、巨噬细胞炎性蛋白1β、IL-8、IL-18 等,从而调节免疫应答[5,13,16-17]。Sun 等[18]研 究 发 现H MGB1 的 增 加 诱 导 了TNF-α和IL-6的第2次释放,表明了某些早期炎性细胞因子与H MGB1之间存在正反馈效应,从而在空间和时间上可进一步放大炎症级联反应。
1.2.2 H MGB1在炎症反应中的时效性 关于HMGB1的时效性目前尚无统一定论。肺脏是脓毒症最易被累及的靶器官,较早并发ALI/ARDS 是死亡的主要原因之一。Wang等[12]用脂多糖 (lipopolysaccharides,LPS)刺激鼠巨噬细胞样RAW264.7细胞株,筛选出了培养基中18 h后明显出现的HMGB1,但HMGB1 m RNA 的表达在16 h内没有上调。说明HMGB1作为一种晚期炎性因子,与早发炎性因子相比具有显著的释放延迟动力学[18]。有学者在LPS诱导小鼠的肺炎症和损伤中发现,诱导后2 h小鼠支气管肺泡灌洗液中HMGB1 水平已能测定,6 h 更高[19]。而有研究发现HMGB1的释放仅在LPS刺激RAW264.7细胞8 h后能被检测到[20]。Chen等[21]使巨噬细胞暴露于烟草烟雾提取物8 h后,可明显观察到核HMGB1易位至胞质和质膜中,至24 h,HMGB1以可溶性分子或以微泡相关的形式释放到胞外。而有学者发现能够抑制HMGB1活性的药物即使在疾病发作后48 h给予小鼠,也可显著提高脓毒症小鼠的存活率[22]。这些研究提示,尽管HMGB1是ALI/ARDS的晚期炎性因子,但它可能会在炎症进程的短时间内表达其毒性。
1.2.3 HMGB1 可作为ALI/ARDS 新的治疗靶点Hatayama等[23]用抗HMGB1单克隆抗体处理ALI小鼠后,小鼠肺组织弥漫性水肿和出血明显减轻。LPS攻击后,巨噬细胞、中性粒细胞及淋巴细胞的数量明显增加,这种增加通过抗HMGB1处理后被显著减弱,而重组HMGB1可提高促炎因子的水平,加剧炎性细胞的浸润[5]。通过诱导HMGB1从胞核易位至胞质,可减少细胞自噬和凋亡,减轻呼吸机所致肺损伤[19]。减少HMGB1 的释放也可减轻ALI的严重程度[24]。故HMGB1 可能是ALI/ARDS 有价值的临床治疗靶标。
2 TLR4与ALI/ARDS
2.1 TLR4的结构、分布与功能 TLR 是具有细胞外配体结合结构域、单个跨膜区段和胞质Toll/IL-1R 结构域的跨膜受体[25-26],其中TLR4基因位于9号染色体,主要在单核细胞、中性粒细胞中表达[27]。TLR4可识别LPS、肺表面活性蛋白A[28]、热休克蛋白70[29]及HMGB1 等配体。其中LPS 刺激TLR4 后,可激活髓样分化因子88(myeloid differentiation factor 88,MyD88)和β 干扰素TIR 结构域衔接蛋白2种途径,这2种途径与其他信号通路的交互可使TLR4信号得到适当调节,最终活化核转录因子κB (nuclear factor-kappaB,NF-κB)和丝裂原活化蛋白激酶 (mitogen-activated protein kinase,MAPK)等信号通路,导致细胞激活或凋亡,以及促炎因子、趋化因子的转录[30-32]。
2.2 TLR4-NF-κB 信 号 通 路 在ALI/ARDS 中 的 作 用LPS、HMGB1等配体与TLR4 结合,通过不同途径激活NF-κB信号通路,诱导下游趋化因子、细胞黏附分子及促炎因子大量释放,募集中性粒细胞,使血管内皮细胞和肺泡上皮细胞受损[5,32-34]。一项脓毒症致大鼠ALI研究中发现,大鼠肺组织中HMGB1、TLR4 和NF-κB 表达明显升高,而在抑制HMGB1-TLR4-NF-κB 信号通路后肺的组织结构和功能有所改善,提示脓毒症所致ALI与此信号通路激活密切相关[35]。Meng等[8]发现在使用右美托咪定干预LPS诱导的ALI后,降低了TNF-α、IL-6和IL-1β的释放,抑制了HMGB1、TLR4、MyD88 和磷酸化NF-κB 通路蛋白的表达,有效减轻了大鼠肺损伤程度。而基因敲除MyD88后可降低TNF-α和NF-κB 的水平,减弱百草枯诱导的小鼠ALI[36]。通过抑制TLR4-NF-κB信号通路,可减轻ALI炎症反应程度。
2.3 TLR4-MAPK 信号通路在ALI/ARDS 中的作用LPS与TLR4结合,激活MAPK 信号通路,导致机体对应激刺激的信号级联反应。已有研究证实p38 MAPK、c-Jun氨基末端激酶 (c-Jun N-terminal kinase,JNK)/应激活化蛋白激酶及细胞外调节蛋白激酶 (extracellular signalregulated kinase,ERK)可能参与了ALI/ARDS 的发展[37],通过调节TLR4-MAPK 信号通路可改善LPS诱导的ALI。木兰花碱可抑制p38 MAPK、JNK 和ERK 活化,从而降低LPS 诱导的ALI中TNF-α、IL-1β和IL-6 的水平[37]。有学者发现丹宁酸抑制LPS诱导的TLR4的表达可能与其抑制了ERK1/2 和p38 MAPK 的活化有关[38]。Wang等[39]在LPS致ALI的体内和体外研究中发现,LPS刺激以时间和浓度依赖性方式增加p38 MAPK 磷酸化,从而导致肺上皮通透性过高和肺水肿,而基因敲除TLR4可消除LPS介导的p38 MAPK 磷酸化,表明TLR4参与LPS诱导的肺上皮屏障功能受损并在p38 MAPK 的上游起作用。有研究发现,p38 MAPK 可能在LPS致NF-κB活化的上游起作用,抑制p38 MAPK 介导的NF-κB信号通路也可成为减轻肺部炎症损害的措施[40]。
3 RAGE与ALI/ARDS
3.1 RAGE的结构、分布与功能 RAGE是免疫球蛋白超家族中的一员,作为一种多配体模式识别受体,与多种炎性疾病的发病有关。人类RAGE编码基因位于6号染色体上。由RAGE基因转录的m RNA 可选择性剪切产生20多种异形体,这些异形体的存在导致多种信号传导胞质衔接子的存在[41]。生理状态下,RAGE在肺上皮细胞中较高基础水平表达,有助于维持肺泡上皮细胞的形态和特定结构,而在巨噬细胞、内皮细胞[42]等其他组织细胞中仅有低水平的表达[43]。病理条件下,RAGE 在内皮细胞[42]、单核细胞[44]等细胞中的表达迅速升高,并与相应配体结合,参与调节炎症反应、细胞凋亡[45]等重要过程。当肺组织受刺激后可表达超生理水平的RAGE、HMGB1[46]和S100蛋白家族[47]等,它们相互作用,介导NF-κB、MAPK、酪氨酸激酶 (Janus kinase,JAK)/信号传导和转录激活蛋白(signal transducer and activator of transcription,STAT)[48]等下游信号通路,产生炎症和氧化应激,影响哮喘、COPD 和ALI等疾病的发生发展[49-50]。
3.2 RAGE-NF-κB 信 号 通 路 在ALI/ARDS 中 的 作 用NF-κB是RAGE下游介导体内炎症反应的重要信号途径之一,且RAGE-NF-κB信号传导的激活可促进RAGE本身的转录,两者之间形成正反馈回路,进一步介导促炎细胞因子分泌[51]。用LPS/HMGB1激活RAW264.7细胞后,新型RAGE拮抗剂降低了细胞表面RAGE 水平,抑制了NF-κB的核转运,同时减少了TNF-α和IL-6的释放,提示体外通过降低RAGE 介导的NF-κB 活化可减轻ALI[52]。在一项用氯胺酮治疗LPS 致大鼠ALI的研究中,氯胺酮下调了RAGE、NF-κB 的mRNA 和 蛋 白 质 水 平,从 而 抑 制 了HMGB-1 mRNA 的上调[46]。这些结果表明,抑制RAGENF-κB信号通路可以作为治疗ALI的新选择。
3.3 RAGE-MAPK 信号通路在ALI/ARDS中的作用 在LPS诱导的ALI小鼠中,通过抑制JNK m RNA 和p38 MAPK 的表达可显著抑制炎症反应[53]。Xiao等[54]的研究结果表明,在体外和体内抑制JNK 磷酸化,可进一步抑制TNF-α、IL-6、IL-1β及COX-2 的表达,表明JNK 在诱导表达炎症介质的上游途径中有调节作用。Li等[55]发现氯胺酮抑制了大鼠ALI中肺组织HMGB1和RAGE的表达,可能部分是由于氯胺酮抑制了MAPK 的激活。且通过抑制RAGE-MAPK 通路可减少氧化应激、抑制自噬并降低炎症反应,改善LPS诱导的ALI[56]。说明抑制RAGE-MAPK信号通路在ALI/ARDS临床治疗中具有潜在价值。
3.4 RAGE-JAK/STAT 信号通路在ALI/ARDS 中的作用 RAGE与HMGB1等配体结合后,可激活JAK/STAT信号转导通路,诱导产生炎性细胞因子[48],可能是ALI/ARDS炎症进程中的一个关键点。有研究发现使用小分子STAT3抑制剂LLL12可减轻炎性细胞浸润,减少IL-1β和CCL2的表达,且LLL12减弱了ARDS患者血浆诱导的人外周血单核细胞中STAT3的磷酸化[57]。另一些药物可通过部分抑制STAT3 转移到胞核[58]、降低JAK1/JAK2-STAT1的总水平和磷酸化水平[59]而在LPS 诱导的小鼠ALI中发挥抗炎作用。而体外下调JAK2/STAT1磷酸化水平并诱导HMGB1从胞核转移至胞质可减少细胞自噬与凋亡[19]。这些发现揭示了RAGE-JAK/STAT 途径与ALI/ARDS的肺部炎症、细胞自噬和凋亡之间的联系。
4 展望
ALI/ARDS发病机制复杂,其中心概念之一是失控的瀑布式炎症级联反应。HMGB1 作为晚期炎性因子,其合成与释放可影响ALI/ARDS的发生发展。HMGB1通过激活TLR4/RAGE等,进一步介导众多下游信号通路,释放多种炎性介质和趋化因子。由于完整的炎症反应可能涉及多种不同但平行的信号途径,故HMGB1 的时效性及TLR4/RAGE介导的多种炎性通路所占比例等尚无明确定论,而该反应涉及到的其他因素也需进一步探索。但目前的研究结果让我们相信,进一步探寻这些分子在ALI/ARDS中的潜在作用机制,可为将来疾病的治疗提供新靶点。
利益冲突所有作者均声明不存在利益冲突
猜你喜欢
中西医结合心脑血管病杂志(2022年19期)2022-11-19
中国现代医生(2022年19期)2022-11-04
中国交通信息化(2022年8期)2022-10-28
中国种业(2022年9期)2022-10-13
中国现代医生(2022年19期)2022-08-25
中国现代医生(2022年21期)2022-08-22
九江学院学报(自然科学版)(2022年2期)2022-07-02
体育科技文献通报(2022年3期)2022-05-23
昆明医科大学学报(2021年12期)2021-12-30
现代临床医学(2021年4期)2021-07-31
