
细胞色素C氧化酶缺乏的可逆性线粒体肌病1例
2021-07-30 01:59唐向国庄家用
儿科药学杂志 2021年8期
唐向国,庄家用
(惠州市第六人民医院,广东惠州 516211)
线粒体肌病是由于线粒体形态和功能不正常所导致的疾病,属少见病,以骨骼肌极度不耐受疲劳为主要特征,病因为线粒体功能缺陷,主要由线粒体DNA(mitochondrial DNA,mtDNA)突变引起,可影响编码线粒体蛋白、转运RNA(transfer RNA,tRNA)、核糖体RNA的相关基因,使线粒体呼吸链氧化磷酸化功能异常,导致三磷酸腺苷(triphosadenine,ATP)合成障碍、能量产生不足而出现的一组多系统疾病[1]。线粒体肌病多在儿童早期发病,病情严重,且进展迅速通常是致命的[2]。但细胞色素C氧化酶(COX)缺乏的可逆性线粒体肌病(reversible COX deficiency myopathy)经早期积极治疗,可完全康复[3],与其他线粒体疾病比较,是目前遗传代谢疾病中唯一可治愈的疾病。现回顾1例经基因检测确诊的COX缺乏的可逆性线粒体肌病患儿的临床资料,总结分析其临床特点。
1 病例资料
患儿,男,2个月2天,因“反复呼吸道感染1月余,反复反应差、发绀11 d”入院。曾在外院诊断“重症肺炎、呼吸衰竭、心肺复苏术后、心肌损害”。给予广谱抗生素、人血丙种球蛋白及呼吸机辅助呼吸等多种治疗,疗效不佳。入院查体:体温36.7 ℃,心率136次/分,呼吸30次/分,体质量3.1 kg,血压70/35 mm Hg,营养中等,气管插管呼吸机通气下血氧饱和度(SpO2)95%以上,肢端暖,毛细血管充盈时间2 s。头颅大小正常,颅缝未闭,前囟0.5 cm。双侧巩膜无黄染,双侧瞳孔等大等圆,对光反射灵敏。口唇无发绀、苍白,颈部对称,无抵抗,双侧呼吸运动对称,呼吸规则,双肺呼吸音粗,闻及固定粗湿啰音,无胸膜摩擦音。心前区无隆起,心尖无抬举感,无震颤,心率136次/分,律齐,各瓣膜区听诊未闻及杂音,无心包摩擦音;腹平坦;肝脾肋下未触及,神经系统查体阴性。患儿系早产,出生体质量2.2 kg,生后诊断“新生儿肺炎、早产儿、心肌损害及低出生体质量儿”。实验室检查:血常规未见明显异常;血气分析中pH 7.212~7.303,二氧化碳分压56.3~76.8 mm Hg,乳酸3.9~7.8 mmol/L;丙氨酸氨基转移酶57 U/L、天冬氨酸氨基转移酶71 U/L;肌酸激酶1 193 U/L,肌酸激酶同工酶155 U/L,乳酸脱氢酶560 U/L;全血丙酮酸3.4 μmol/L;肌钙蛋白T 0.169 ng/mL;肺炎支原体、衣原体IgM阴性;痰培养示铜绿假单胞菌;胸片示支气管肺炎;头颅磁共振成像(MRI)示脑白质髓鞘化进程相当于1月龄婴儿水平,余脑部MRI未见明显异常。
患儿入院后予呼吸机辅助通气,哌拉西林/舒巴坦抗感染,人血丙种球蛋白、白蛋白及雾化吸入等对症治疗。患儿脱机后,入院第23天鼻导管吸氧下突然出现心肺骤停,给予心肺复苏后自主呼吸恢复及再次呼吸机辅助通气,复查动脉血气分析示二氧化碳潴留及血乳酸持续升高,床边胸片未见明显异常。考虑线粒体脑肌病,给予线粒体鸡尾酒疗法(辅酶Q10、左卡尼丁及三磷酸腺苷),并调整为头孢他啶注射剂抗感染。第28天患儿再脱机,继续予线粒体鸡尾酒疗法及抗感染。第34天患儿病情平稳,低流量吸氧(家长自购制氧机)可维持,无明显气促,因无法自行进食,携带胃管出院。出院后随访,预后良好。
2 基因检测
为确诊病因,经患儿家属知情同意后,采用乙二胺四乙酸(ethylene diamine tetraacetic acid,EDTA)抗凝管抽取患儿及父母外周静脉血各4 mL,送北京金准基因科技有限公司金准医学检验所检测。1个月后基因检测报告结果显示,线粒体基因组(chrM)14674位点突变,家系验证结果显示此突变来自其母亲。见图1。金准点评显示,COX缺乏的可逆性线粒体肌病是由于母系遗传,同质性m.14674 T>C突变导致的疾病,几乎全部患儿在出生数周内都具有严重的肌张力减退、呼吸和进食困难及血清乳酸浓度升高。若患儿在出生数月后存活,此疾病会表现出完全(或几乎完全)的自发性恢复,5~20个月自发改善,且通常在2~3岁时恢复正常。

患儿线粒体chrM:14674存在T>C的突变

患儿母亲线粒体chrM:14674存在T>C突变
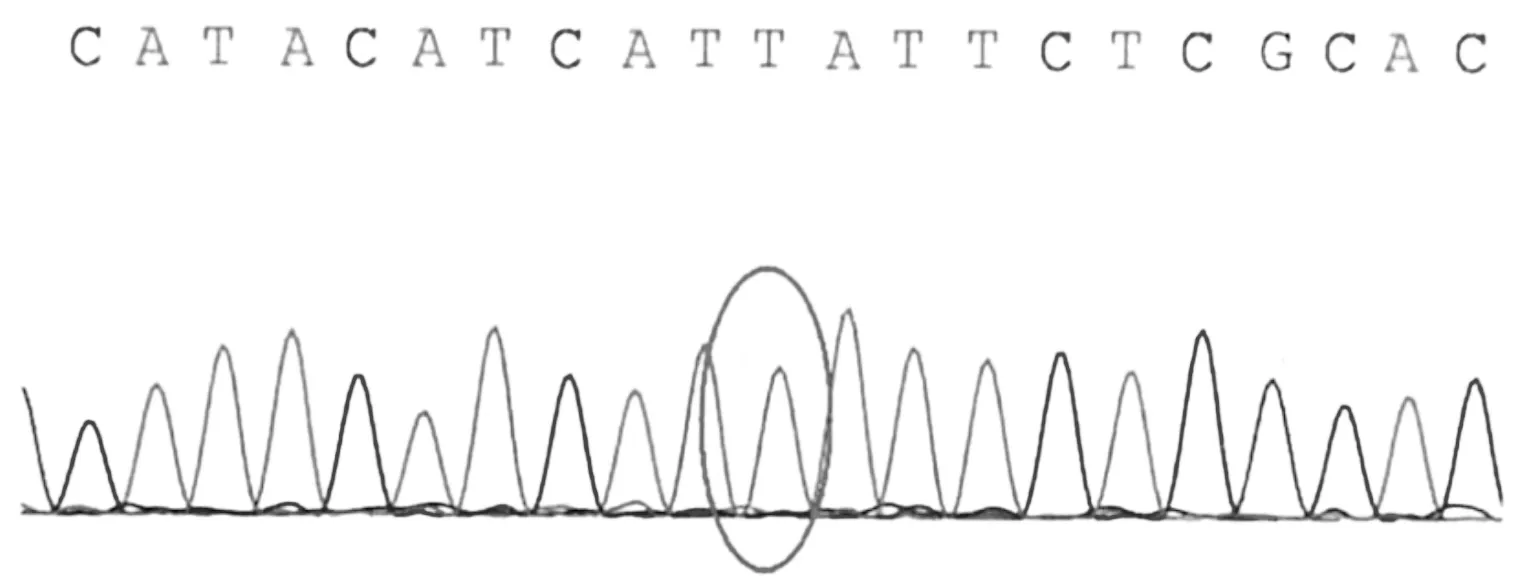
患儿父亲线粒体chrM:14674无突变
3 讨论
线粒体肌病多在婴幼儿发病,病情严重,且进展迅速,目前缺乏彻底有效的治疗方法,预后不良。但COX缺乏的可逆性线粒体肌病是目前遗传代谢疾病中唯一发现可治愈的疾病[4]。Van Biervliet J P等[5]1977年报道1例严重乳酸酸中毒伴肾病、脑损伤患儿,所有治疗方法均无效,至4个月时死于呼吸衰竭,其同胞死于类似症状,肌线粒体生化检查示COX缺陷。DiMauro S等[6]在1981年报道1例男婴,消瘦、软弱、垂睑,无力吸奶及有吸入性肺炎,严重血乳酸酸中毒,生长发育明显落后,肌活检标本的生化学分析结果显示COX活性减至约正常值的8%,随年龄增长(14个月后),血乳酸降至正常水平,临床症状明显改善,其COX活性自动好转,但具体原因未明,称之为COX缺乏的可逆性线粒体肌病。此后,越来越多的类似病例被报道。在2009年,Horvath R等[7]在婴儿起病COX缺乏的可逆性线粒体肌病的患儿中发现同质性m.14674 T>C基因突变,为mtDNA编码谷氨酸tRNA基因(mt-tRNAGlu)的点突变,损害mtDNA翻译,影响转运谷氨酸(Glu)的功能,可造成几种关于谷氨酸-rRNA的蛋白质合成缺陷,其参与执行氧化磷酸化功能蛋白的组装,导致COX缺乏。由于基因突变为病因,开启基因检测诊断婴儿线粒体肌病及预后评估[8]。
COX缺乏的可逆性线粒体肌病的临床特征表现为在婴儿期临床症状自发性改善、COX缺乏可逆性改变。目前COX缺乏的可逆性线粒体肌病的发生机制尚不明确,但Boczonadi V等[9]在2015年报道COX亚单位可逆性的改变,有助于该病发生机制的解释。机体的不同组织细胞因代谢需精细调节COX活性,即细胞核编码的COX亚单位(COX6A、COX7A)为组织特异的基因表达。心脏、骨骼肌中基因表达COX6A、COX7A的水平不同于肝脏、肾脏及大脑。肝脏COX6A、COX7A基因表达出现早(生后即刻)、表达量大,而心脏及骨骼肌出现晚(约生后3个月)、表达量缓慢增加,并持续终生。COX6A、COX7A呈年龄依赖生理性改变趋势,即随年龄增长(生后3个月),COX活性自动完善(可逆性)。
Salo M K等[10]在1992年报道父母非近亲婚配的 2例患儿,生后不久出现吸吮、呼吸困难及肌无力,肝肿大,血肌酸激酶异常增高及乳酸酸中毒,肌活检电镜下的线粒体明显肿胀,第一次活检标本生化学分析结果示COX活性减至约正常值的25%,均予鼻饲喂养,男性患儿3岁和女性患儿28个月时已无临床表现。Wada H等[11]在1996年也报道了1例2个月女性患儿,软弱、肌张力降低、肝肿大及严重血乳酸酸中毒,需机械通气支持,肌活检标本生化学分析结果示COX活性减至约正常值的16%,予辅酶Q10、左卡尼丁治疗,血乳酸持续降低,临床症状好转,8个月后其COX活性恢复正常,辅酶Q10、左卡尼丁治疗有效,早期诊断及强化治疗有利于临床症状的改善。本例患儿因呼吸困难起病,血清肌酸激酶、血乳酸异常增高,头颅MRI示脑白质发育落后,早期积极强化治疗(呼吸机辅助通气改善通气等),予辅酶Q10、左卡尼丁等(线粒体鸡尾酒疗法),临床症状明显改善,预后良好。基因检测示线粒体chrM-14674 T>C基因突变,为COX缺乏的可逆性线粒体肌病,属于良性进展过程,早期予ATP,代偿能量不足,渡过危险期,随年龄增大(约生后3个月),COX活性逆转,临床症状逐渐消失。
COX缺乏的可逆性线粒体肌病为母系遗传方式[12],母亲将mtDNA传递给子女,只有女儿将其mtDNA传递给下一代,但不遵循孟德尔定律,因mtDNA突变是随机分配到细胞中,是否起病取决于mtDNA突变体占mtDNA总数的比例,通常mtDNA突变体低于40%不发病,即不是所有基因突变者均发病。因此,母亲可为携带者,而子女是否患病、患病轻重程度取决于获得母亲mtDNA突变体的数量。本例患儿及其母亲检测线粒体基因组为同样的基因突变,而其父亲无,家系验证结果示此突变来自其母亲,但其母亲无临床症状(携带者)。
综合上述,COX缺乏的可逆性线粒体肌病是目前遗传代谢疾病中唯一可治愈的线粒体疾病,不同于其他类型的线粒体病,关键在于早期诊断,临床发现血乳酸、肌酶持续增高,且其他原因不能解释,尤其是合并脑发育不良时,需考虑线粒体病的可能,尽早基因检测明确诊断,如示chrM14674 T>C突变,则为良性进展过程,经早期积极治疗,预后良好。
猜你喜欢
大学图书馆学报(2023年1期)2023-02-15
今日农业(2021年5期)2021-11-27
中国医学影像学杂志(2021年6期)2021-08-13
现代畜牧科技(2021年5期)2021-07-20
中华养生保健(2020年8期)2021-01-14
成都工业学院学报(2018年3期)2018-05-14
腹腔镜外科杂志(2016年12期)2016-06-01
兽医导刊(2016年6期)2016-05-17
恋爱婚姻家庭·养生版(2016年2期)2016-02-17
河北科技大学学报(2015年6期)2015-03-11
