
黑磷纳米光催化材料研究进展
2021-09-18 08:25沈志凯元勇军于振涛邹志刚
中国材料进展 2021年7期
沈志凯,元勇军,于振涛,邹志刚
(1. 杭州电子科技大学材料与环境工程学院 新能源材料研究中心,浙江 杭州 310018) (2. 南京大学现代工程与应用科学学院 固体微结构物理国家重点实验室,江苏 南京 210093)
1 前 言
2019年,全球二氧化碳(CO2)排放量高达368亿吨,其中95%以上是由化石能源使用产生的[1, 2]。化石能源的大量使用不仅造成不可再生资源的减少,同时带来了严峻的环境问题与全球气候变化。目前,全球主要经济体(如欧洲、中国、日本等)都已经制定了碳中和目标,我国确认将于2030年前后达到CO2排放峰值,并力争在2060年前实现碳中和的目标,届时工业领域将以电能为主,氢能为辅[3-5]。这一碳达峰碳中和计划不仅要求减少化石能源的使用,大力推广清洁能源,还要求对已经造成的环境问题进行修复。
光催化技术,包括光催化制氢(photocatalytic hydrogen production,HER)、人工固氮(photocatalytic nitrogen reduction,NRR)和CO2还原等,通过太阳能驱动水分解、N2还原和CO2还原反应,为清洁氢能制备、低能耗固氮和CO2再利用提供了一种新的技术途径。因此,光催化技术对实现碳中和碳循环具有重要意义。基于半导体材料的光催化过程可以分为以下几步:材料被能量大于或等于其禁带宽度(Eg)的光照射后,产生光生电子-空穴对,电子跃迁到导带(CB),价带(VB)出现空穴,电子和空穴分别在材料的表面发生还原反应与氧化反应[6]。此外,半导体催化剂的导带位置应该比水/氮/CO2的还原电位更负,才能发生相应的还原反应[7],不同反应的方程式及相应的还原电位如表1所示。另一方面,作为光催化剂还应该有广泛的光吸收范围、丰富的表面活性位点、高载流子迁移率等特点。目前可用的光催化材料十分丰富,覆盖不同的类型,如金属氧化物TiO2[8]、Fe2O3[9]、WO3[10]、BiVO4[11]等;金属硫化物CdS[12]、MoS2[13]、WS2[14]等;碳基材料石墨烯[15]、g-C3N4[16]等;以及无机化合物MXene[17],金属-有机框架材料MOF[18]等。即使已经有如此多材料被研究用于光催化,仍然很少有材料能同时满足产氢、固氮和CO2还原的要求,又兼备广泛的光吸收范围,因此,找到同时满足上述条件的光催化材料对于光催化的发展具有十分重要的意义。
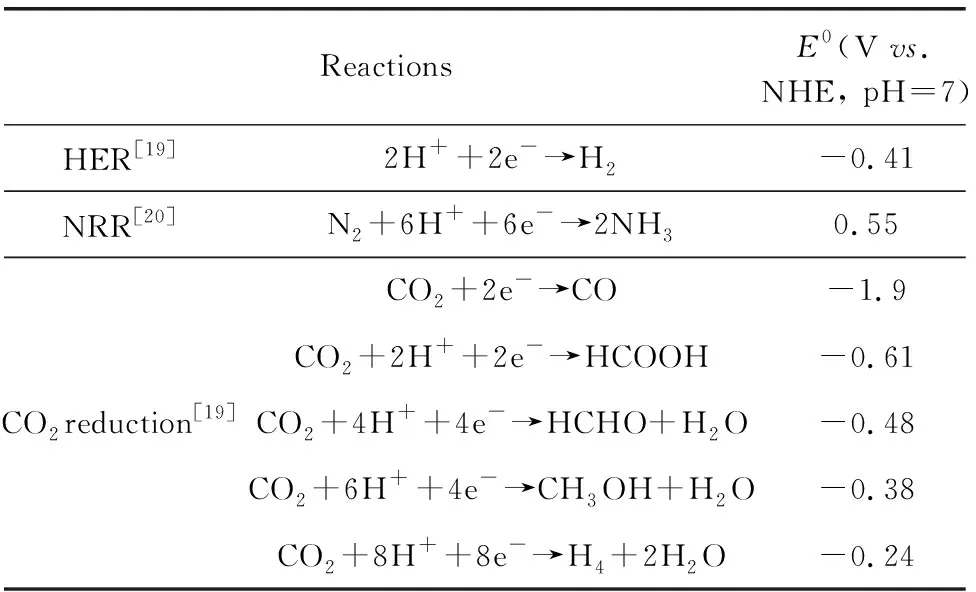
表1 不同光催化的反应方程式和还原电位
黑磷材料作为一种新兴的二维材料,近几年受到了研究人员极大的关注[21]。黑磷晶体由一层层的片状结构堆叠而成,层与层之间通过弱的范德华力结合,因此可以通过剥离形成二维(2D)结构[22]。相比于其它二维材料,黑磷材料具有诸多独特的优点。黑磷具有带隙可调节性,从块体材料的0.3 eV变化到单层纳米片的2.0 eV,吸收范围可覆盖紫外-可见-近红外区域。更重要的是,黑磷具有高电子迁移率,有利于载流子的传导,促进电子-空穴对的分离,从而提高催化性能。基于上述特性,黑磷材料已经在光催化制氢、人工固氮和CO2还原等领域被广泛研究和应用[23]。
本文从介绍黑磷材料的结构与特性出发,总结了黑磷纳米材料的制备方法,聚焦黑磷材料在光催化上的应用,包括用于光催化产氢、固氮、CO2还原和有机物降解等方面,结合相关最新的研究进展与案例,讨论了黑磷材料在光催化中的应用现状。
2 黑磷材料的特性
黑磷,作为磷元素的一种存在形式,是红磷与白磷的同素异形体[24]。少层黑磷纳米片表现出很多独特的性能,比如大的比表面积、高载流子迁移率、出色的开/关比、面内各向异性,以及基于层数可调节的禁带宽度[25]。正是这些优异的特性,使得黑磷材料在很多方面都是理想的材料。但是黑磷材料也存在一定的缺点,其很容易在空气与水中被氧化而降解,具有较低的化学稳定性。
2.1 黑磷的晶体结构
如图1所示,黑磷晶体由单层的黑磷堆叠而成。根据层数及尺寸,可以将黑磷分为量子点、单层、少层、块体这几种不同的类型。黑磷层与层之间通过较弱的范德华力连接,层间距在3.21~3.73 Å之间[26],因此可以通过块体晶体剥离得到尺寸更小的黑磷纳米材料。在黑磷晶体结构中,磷原子的3p轨道上有5个价电子,原子与原子之间形成sp3杂化,每一个磷原子与两个面内原子和一个相邻平面内原子共价连接而饱和[27]。沿Armchair(AC)方向具有褶皱结构,沿Zigzag(ZZ)方向表现为双层结构。面内原子(Armchair方向)之间的键角为94.1°,键长2.253 Å,键角会随层数减少略有变小;Zigzag方向原子之间的键角为103.3°,键长2.287 Å,键角会随层数减少略有变大[28],但层数对键长几乎没有影响[29]。
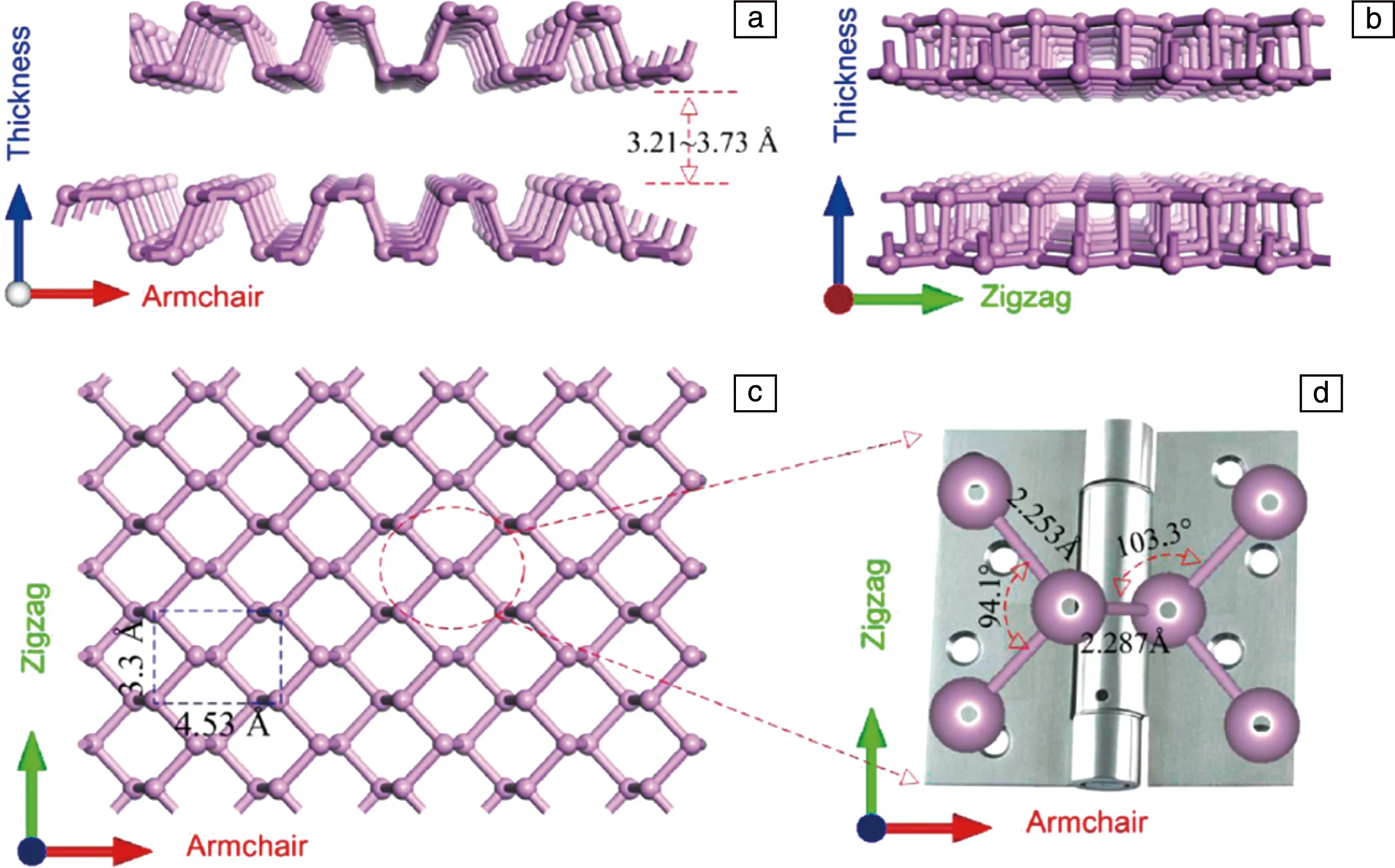
图1 黑磷的结构示意图[26]:(a)Armchair方向侧视图,(b)Zigzag方向的侧视图,(c)顶视图,(d)P—P键的局部放大图Fig.1 Structure of black phosphorene(BP)[26]: side views from the Armchair (a) and the Zigzag (b) direction; (c) top view; (d) zoomed-in local atomic structure of the P—P bonding configuration
2.2 黑磷的可调带隙
黑磷属于直接带隙半导体,且该特性与层数及尺寸无关[30]。但是黑磷的禁带宽度受到层数影响,这是黑磷材料的一大特性。随着黑磷层数的减小,其禁带宽度会逐渐变大。2014年,Cai等通过第一性理论计算,详细研究了少层黑磷中禁带宽度与层数之间的关系[31]。发现随着层数的变化,价带顶位置的变化幅度比导带底的大(图2a)。黑磷作为一种p-型半导体,价带顶灵敏地关于层数调整的特性,有助于调整肖特基能垒的高度,从而更有效地促进电荷传输过程。除了层数之外,掺杂[32]、外部电场[33]、化学功能化[34]和应变(压缩或膨胀应变)[35]也可以调节黑磷的禁带宽度。基于黑磷材料禁带宽度出色的可调性,研究人员可以通过调节厚度获得不同带隙的材料,而不同带隙的材料具有不同的光吸收特性。通过调控,黑磷材料光吸收范围可以覆盖紫外-可见-近红外区域,调节范围远大于其他半导体材料[36]。填补了石墨烯(接近于零带隙的半导体)与过渡金属二卤化物(宽带隙的半导体)光吸收范围之间的空白部分,这是黑磷在光催化领域得到广泛研究的重要原因之一。
2.3 黑磷的稳定性

研究人员从实验与理论等多个角度研究了黑磷在环境中降解的机理。结果表明,黑磷易于降解的特性与磷原子的孤对自由电子有关,未完全成键的边缘磷原子可能会产生不稳定的结合结构[38]。2015年,Ziletti等的第一性原理计算结果表明,黑磷表面与氧气反应是其降解的首要原因,氧原子的三重-单峰转化极大地降低了解离势垒,而与磷结合后的氧原子又提高了亲水性,会进一步加速降解[39]。同年,Favron等通过实验证明了降解机理是黑磷与溶解在水中的氧气发生了光诱导氧化反应,氧气、水和可见光都是黑磷氧化的因素,并且氧浓度和光强度可以线性地影响黑磷的降解速率[40]。
3 黑磷纳米材料的制备方法
3.1 黑磷晶体的制备方法
黑磷晶体的合成可以追溯到一百多年前。1914年,Bridgman等首次通过高压法合成黑磷晶体:以白磷为原料,在1.2 GPa与200 ℃下保持5~30 min,即可将白磷转变为黑磷[41]。1981年,Shirotani首先使用楔型立方高压装置,在高温高压(3.8 GPa和高于270 ℃)下,将红磷转变为黑磷单晶[42],但如此严苛的合成条件极大阻碍了对于黑磷的研究。1989年,Maruyama等通过铋-汞熔法合成了针/棒状的黑磷,将白磷用15%HNO3纯化,然后溶解在液态铋中,加热到400 ℃后保温20 h,缓慢冷却到室温后,用30%的HNO3溶解铋,制备得到黑磷晶体[43]。目前,实验室使用最多的合成方法是矿化法。这种方法原材料简单,可在低温低压下制备。合成过程中以红磷为原料,碘化硒作为矿化剂,添加金[44]、铜[45]或硒[24]等材料作为催化剂,混合后在低压下加热,在650 ℃下保温6 h,用甲苯和丙酮清洗并真空干燥后即可获得黑磷晶体。目前,无论哪种方法,黑磷的产率都比较低,如何提高黑磷的产率以及纯度,是未来黑磷合成方法改进的重点[46]。
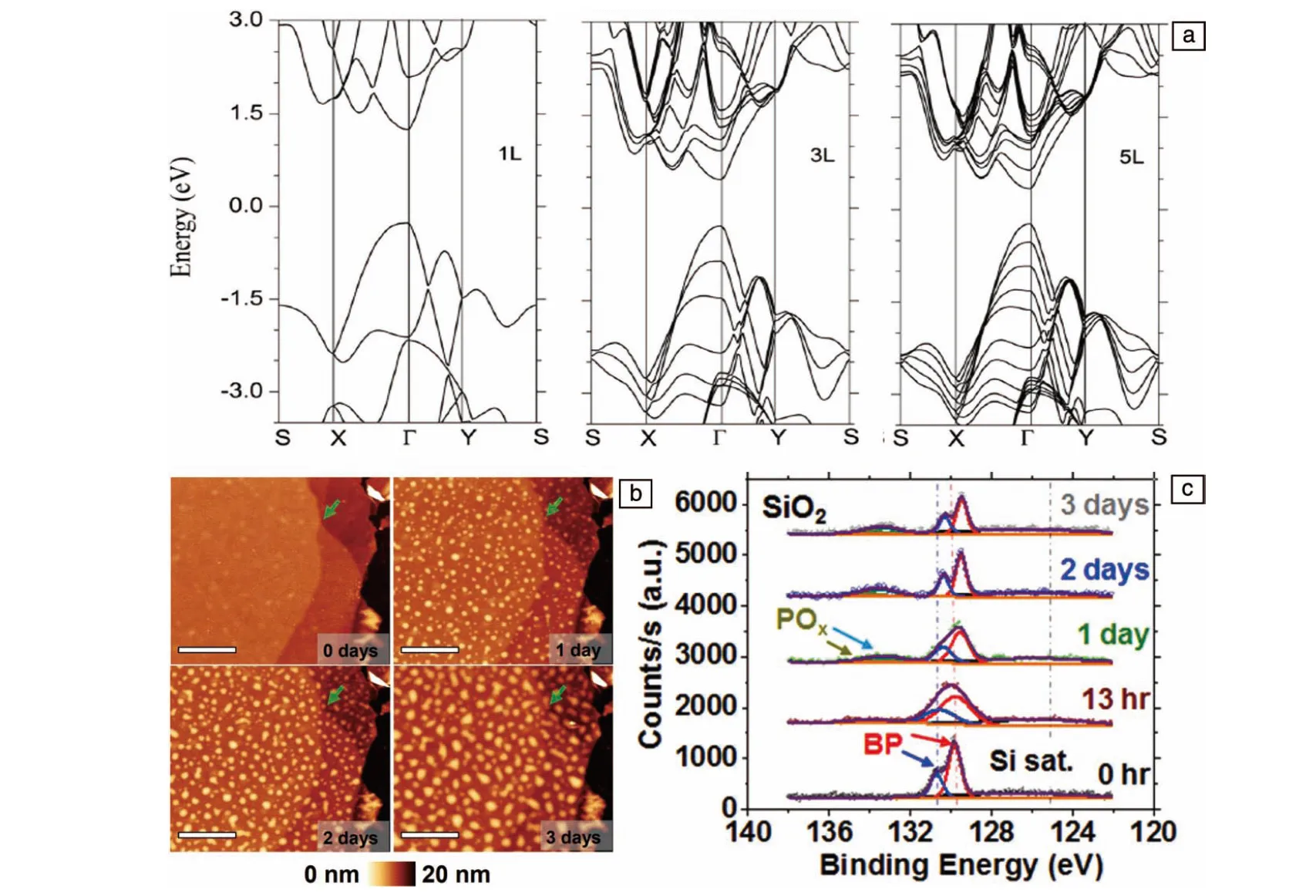
图2 由HSE06方式计算获得的不同层数黑磷的能带结构(a)[31];黑磷剥离后暴露于空气中不同时间后的AFM照片(b)和XPS图谱(c)[37]Fig.2 Band structures of different layered BP by HSE06 calculation(a)[31]; AFM images(b) and XPS spectra (c) of BP after being exposed to air for different time[37]
3.2 黑磷纳米片的制备方法
与其它二维材料相似,黑磷纳米片的制备方法主要有两种:自上而下剥离法和自下而上合成法。其中,自上而下剥离法是目前使用最多的方法,包括胶带法、机械剥离法、液相超声法和电化学剥离法等,其特点是需要先合成块体黑磷晶体,而后进行剥离得到纳米片。自下而上合成法包括溶剂热法、化学气相沉积法等。相比于前者,这些方法操作步骤更少,且合成温度更低。上述方法各有特点,适用的领域也有所不同,本文介绍几种在催化应用上使用比较多的制备方法。
3.2.1 液相超声法
液相超声法通过将黑磷晶体材料分散在溶液中,超声一定时间后,经过不同转速的离心分离出所需尺寸的黑磷纳米片[47]。在超声过程中,由于波动和剪切力产生的微米和纳米级气泡与溶剂分子一起通过插层扩展了块体黑磷,从而克服了黑磷层之间的范德华力,将黑磷纳米片分散到溶液中[48]。其中,选择合适的溶液是剥离的关键。2014年,Brent等通过使用有机溶剂N-甲基吡咯烷酮(NMP)为溶液,将块体黑磷置于其中冰浴超声24 h,最后通过高速离心得到黑磷纳米片[49]。同年,Yasaei等系统地实验研究了N,N-二甲基甲酰胺(DMF)与二甲基亚砜(DMSO)对黑磷的剥离效果(图3a和3b),发现使用DMF更容易制备尺寸更小的黑磷纳米材料[50]。同时研究人员认为通过该方法制备的黑磷纳米片拥有与机械剥离法相似的电性能。此外,该研究还发现超声功率与时间也是剥离效率的重要影响因素。2015年,Sersht等通过理论计算对比了不同溶液对黑磷剥离效率的影响[51],表明剥离效率取决于溶剂与材料表面之间的能量匹配程度,过高或过低都会降低剥离效率。同年,Guo等通过在NMP中加入氢氧化钠作为辅助剂,提高了黑磷的剥离效率和稳定性[52]。到目前为止,使用NMP作为溶剂的液相超声法依然是剥离黑磷纳米片的最主要方法,但这一高沸点的溶剂却难以从黑磷纳米片表面去除干净,限制了黑磷的应用。为此,Wang等使用无氧去离子水对黑磷进行超声处理(图3c),通过延长超声时间与提高功率得到黑磷纳米片[53]。此外,离子液体[32],表面活性剂[54]也被用于黑磷纳米片的剥离。
3.2.2 电化学插层剥离
将块体黑磷固定在电极夹上作为工作电极,在其与对电极之间施加一定的电位,溶液中的离子插入到黑磷的层间,在电场作用下产生体积膨胀,从而克服层间的范德华力,得到单层/少层的黑磷纳米片(图3d和3e)。在此过程中,离子的大小是关键,过小无法产生足够的膨胀,过大则无法进入层间[55]。2018年,Yang等以四正丁基硫酸氢铵(TBA·HSO4)为电解质,无水脱氧碳酸亚丙酯为溶液,施加-8.0 V的恒定电位成功得到黑磷纳米片[56]。作为对比,其它无机盐以及季铵盐也用于研究,如高氯酸锂(LiClO4),高氯酸钠(NaClO4),四甲基硫酸氢铵(TMA·HSO4)以及四乙基硫酸氢铵(TEA·HSO4)等,这些离子的剥离效率如图3f所示。由于正丁基链的良好柔韧性以及合适的尺寸,在使用四正丁基硫酸氢铵时剥离效率达到最大值78%,同时最大尺寸达到20.6 μm。另外,Kovalska等还探索了在低电位下的电化学剥离,在含烷基铵离子的无水无氧低沸点乙腈(AN)和DMF溶液中,仅分别施加-2.95和-2.85 V电压即可开始剥离,快速剥离只需要-3.8 V[57]。除了上述非水性溶剂,2017年,Pumera等使用0.5mol/L的稀硫酸(H2SO4)作为溶液,在施加-3 V电压下,也成功制备得到黑磷纳米片[58]。2021年,Wang等用电化学插层法制备出了大尺寸的黑磷纳米片。以DMF为溶液,弱酸性的四正丁基乙酸铵(CH3COOTBA)为溶质进行电化学插层,获得的黑磷纳米片的平均尺寸高达(77.6±15.0) μm,最大尺寸可以达到119 μm。其原因是TBA+与配位的弱路易斯酸之间的相互作用较弱,从而阻碍了氢还原反应,提高了剥离率,且所得纳米片的尺寸随H+或H2O含量的提升而减小[59]。
3.2.3 溶剂热合成法
2018年,Tian等首次基于自下而上法合成出黑磷纳米片,如图3g所示[60]。以白磷作为前驱体,乙二胺为溶剂,在一定温度下反应,冷却后清洗并真空干燥。在此过程中,溶剂的选择和反应温度是决定黑磷纳米片的形成及厚度的关键因素,通过实验发现,在60~140 ℃范围内都可以合成黑磷纳米片,并且随着温度的升高产率也越高。所得纳米片的尺寸约为800~1000 nm,厚度在15 nm以下。2020年,Sun等也通过相似的方法,制备得到黑磷纳米片,并研究了溶液对合成黑磷纳米片催化性能的影响。他们以红磷为前驱体,分别以乙二胺(eda-BP),1,3-丙二胺(pda-BP)和1,4-丁二胺(bda-BP)为溶液,在160 ℃下反应12 h,并且使用液相超声法剥离制备了对比样品(ctv-BP),最终将所得的黑磷纳米片用于光催化产氢。实验结果(图3h)表明,以乙二胺为溶液反应得到的黑磷纳米片表现出最高的产氢活性[61]。同年,Zhu等使用红磷为前驱体、乙二胺为溶剂,探究了不同温度对合成的黑磷纳米片的光催化产氢性能的影响,证明在165 ℃下反应24 h得到的材料催化性能最好[62]。溶剂热合成法从另一方向拓展了黑磷纳米片的制备方式,且具有操作简单的优点。但与其他方法相比,目前也存在一定的缺点,比如合成的黑磷纳米片结晶性比较差且容易被氧化。
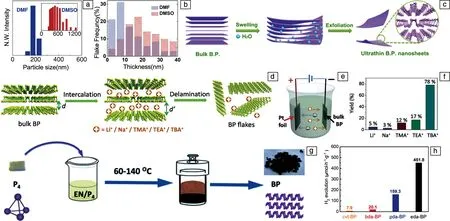
图3 通过DMF和DMSO剥离得到的黑磷的尺寸分布图(a)和厚度分布图(b)[50];水作为溶剂液相超声法剥离黑磷纳米片的示意图(c)[53];电化学膨胀法剥离黑磷纳米片的原理图(d)和装置示意图(e);在电化学膨胀法中使用不同无机盐及季铵盐的剥离效率(f)[56];溶剂热法合成黑磷纳米片的示意图(g)[60];使用不同溶剂剥离得到的黑磷纳米片的光催化产氢性能(h)[61]Fig.3 Size distribution graph (a) and thickness distribution graph (b) of black phosphorus obtained in DMF and DMSO[50]; schematic diagram of preparing black phosphorous nanosheets by liquid phase ultrasonic method using water as solvent (c)[53]; illustration of the delamination procedure and reaction cell (d, e); the efficiency of using different inorganics in the electrochemical expansion method (f)[56]; schematic diagram of solvothermal synthesis for black phosphorous nanosheets (g)[60], photocatalytic hydrogen production performance of black phosphorous nanosheets obtained in different solvents (h)[61]
3.3 黑磷量子点(BPQDs)的制备方法
黑磷量子点(BPQDs)的制备方法大多与黑磷纳米片的制备方法一致,比如机械剥离法、液相超声法等,在此基础上通过改变部分实验条件参数即可剥离得到量子点。除此之外,还有微波辅助剥离法,相比于液相超声法,其所需时间更短。溶剂热法既可以自下而上合成出黑磷晶体,也可以以块体黑磷晶体为原料,剥离得到黑磷纳米片或量子点。目前,很多研究人员也在探索新型制备方法,如搅拌机破碎法[63]、脉冲激光法[64]等。
2014年,Sun等首次通过液相超声法制备得到黑磷量子点。将黑磷晶体分散在少量NMP溶液中,并在研钵中研磨20 min,然后加入适量NMP溶液,并在冰浴下连续超声3 h,最后以7000 r/min离心20 min,即可得到黑磷量子点,其尺寸在4.9 nm左右,厚度1.9 nm[65]。在液相超声中,共有2种超声方式:尖端超声与连续超声。两种方式均可将块体黑磷晶体剥离至更小的状态,区别是单独使用液相超声会更多剥离成纳米片,而不是量子点。尖端超声可以直接使用块体黑磷进行剥离,而不需要先磨成粉末状[66]。2017年,Batmunkh等通过微波辅助/液相剥离制备得到黑磷纳米片,将黑磷晶体分散在NMP溶液中,使用微波加热至50 ℃并保持10 min,经过离心即可得到黑磷纳米片[67]。2019年,该团队将微波加热温度和时间分别提高到120 ℃和30 min,成功使用该方法制备出黑磷量子点,所得黑磷量子点的平均尺寸在2.95 nm左右[68]。此外,2018年,Long等将液相超声法与溶剂热法相结合,以乙醇为溶剂制备出黑磷量子点[69]。首先将黑磷晶体分散在乙醇中,超声6 h后以8000 r/min离心10 min以去除未剥离的部分,然后将上层液体在80 ℃下溶剂热反应4 h,通过在40 ℃下蒸发乙醇后得到黑磷量子点,平均尺寸在2.5 nm左右。
4 黑磷材料在光催化中的应用
4.1 光催化产氢
目前光催化领域内研究较多的材料,如g-C3N4、CdS、TiO2等,它们的光吸收范围主要位于紫外光区和部分可见光区,而这在太阳光中只占极小部分。黑磷材料由于其出色的带隙调节性能,光吸收范围可以覆盖紫外到近红外区域,从而提高太阳光的利用率。2019年,Majima等将黑磷、氧化石墨烯(GO)与MoS2相结合,用于可见-近红外光下的光催化产氢[70]。制备方法如图4a所示,将单层GO与块体黑磷晶体在NMP中超声,通过离心去除大尺寸材料,再加入MoS2共同超声搅拌,得到MoS2-BP/GO材料。黑磷因其出色的光吸收性能作为光催化剂,MoS2作为助催化剂,而GO一方面用作表面活性剂促进BP的剥离,另一方面其局部共轭的芳族体系和良好的电子穿梭能力,促进了光催化反应期间有效的电荷分离/转移。UV-vis吸收光谱(图4b)表明MoS2纳米片的光吸收范围主要覆盖可见光区,BP/GO在紫外到近红外区域表现出广泛的吸收,加入MoS2形成MoS2-BP/GO后,吸收性能进一步提升,表现出在可见-近红外区光催化的潜力。对材料在350~1800 nm下进行光催化产氢测试3 h,结果表明(图4c),纯黑磷与MoS2没有催化活性,MoS2-BP/GO表现出最高的活性。在催化过程中,黑磷被光激发产生电子-空穴对,GO作为黑磷与MoS2之间的连接材料,由于其良好的载流子迁移率,可以将黑磷导带的电子传输到具有更低功函数的MoS2的导带,在此发生还原反应生成H2,而在黑磷的价带,空穴与作为牺牲剂的甲醇反应而消耗,从而达到在可见-近红外光区的有效光催化产氢。此外,贵金属材料也可用来提升催化性能,通过在黑磷纳米片上负载Pt,得到Pt/BP异质结催化剂,展现出出色的稳定性和广泛的吸收范围,可以覆盖到1800 nm以上的红外区域[71]。
2017年,Zhu等首次报道了黑磷纳米片在水中进行可见光催化产氢的应用[35]。在LiOH的辅助下,对黑磷晶体进行球磨得到黑磷纳米片,在没有任何助催化剂的情况下,光催化性能达到512 μmol·h-1·g-1,大约是块体黑磷晶体的18倍,表现与g-C3N4相当。为了提升黑磷光催化产氢性能,Moon等通过引入Ni2P颗粒作为助催化剂[72],提升载流子分离效率,从而减少光生载流子的复合,提高了黑磷的光催化产氢性能。Ni2P作为助催化剂,是反应的活性中心。黑磷纳米片巨大的表面积为其负载提供了大量的空间,同时有利于暴露更多活性位点。此外,黑磷的高电导率有效地将电荷载体转移到光催化剂表面并增强了光催化活性。光催化产氢性能达到858.2 μmol·h-1·g-1。但是颗粒材料容易发生团聚而降低催化性能,针对这一问题,研究人员尝试将两种二维材料复合,形成2D-2D异质结。
2018年,Hu等将二维黑磷纳米片与二维Bi2WO6(MBWO)结合,形成2D-2D接触的BP/MBWO异质结催化剂[73]。通过引入溴代十六烷基三甲胺(CTAB)改变合成条件,传统花状的Bi2WO6会转变成片状形貌,将Bi2WO6纳米片与液相剥离得到的黑磷纳米片于NMP中混合,经过超声与搅拌,得到BP/MBWO异质结材料。由SEM照片(图4d)可知,BP均匀分散附着在MBWO表面,两者之间形成紧密接触。为了了解其光学性质,进行UV-vis吸收光谱测试。MBWO的吸收边大约在450 nm,在加入BP之后,这一点并没有改变。但是在可见光区域的吸收却有了很大的变化,随着BP含量的提升,在400~800 nm内的吸光强度逐步增强。这证明黑磷的加入可以显著提升材料对可见光的吸收。在添加质量分数3% H2PtCl6·6H2O作为助催化剂的情况下,对这些材料进行光催化产氢测试。结果表明,加入BP后,MBWO的催化性能明显提高,并在含量为12%时表现出最高的性能:21 042 μmol·g-1(5h)。其光催化机理是,BP与MBWO形成Z型异质结,MBWO导带的电子与BP价带的空穴复合,从而提高载流子分离效率。BP导带中的部分光生电子迅速移动到Pt并将H+还原成H2。2019年,作者团队将黑磷纳米片与MoS2结合,形成2D-2D异质结[74],认为2D-2D的接触机制显著增大了两种材料的接触面积,从而增加了载流子迁移通道,进而提高载流子分离效率,提升光催化产氢性能,催化活性远高于负载3%Pt(质量分数)的黑磷纳米片(0D-2D异质结)。此外BP/MoS2材料也表现出良好的稳定性,其原因可能是MoS2大面积地覆盖在BP表面,一定程度上阻碍了BP与环境的接触,减缓了降解,从而提升稳定性。
除了黑磷纳米片,黑磷量子点在光催化产氢中也有很多研究。2018年,Cai等将黑磷量子点、Au纳米棒和CdS纳米线相结合形成异质结,在以Na2S/Na2SO3为牺牲剂的情况下,实现了全光谱下的光催化产氢,并对BPQDs在光催化中电荷分离情况进行了首次深入研究[75]。从图4e可知,BP-Au-CdS三元异质结表现出最优的催化性能,为10.1 mmol·h-1·g-1,显著高于二元异质结材料。此外,在近红外区(λ>700 nm)的催化活性达到了4.3 mmol·h-1·g-1,展现了全光谱下光催化产氢活性。分析其催化机制(图4f), BP-Au-CdS被光照后,BPQDs产生光生电子-空穴对,其导带电子可以转移到Au纳米棒上;与此同时,Au纳米棒因表面等离子体共振效应(SPR)也会产生电子和空穴,电子可以单向转移到CdS的导带,还原水产氢。
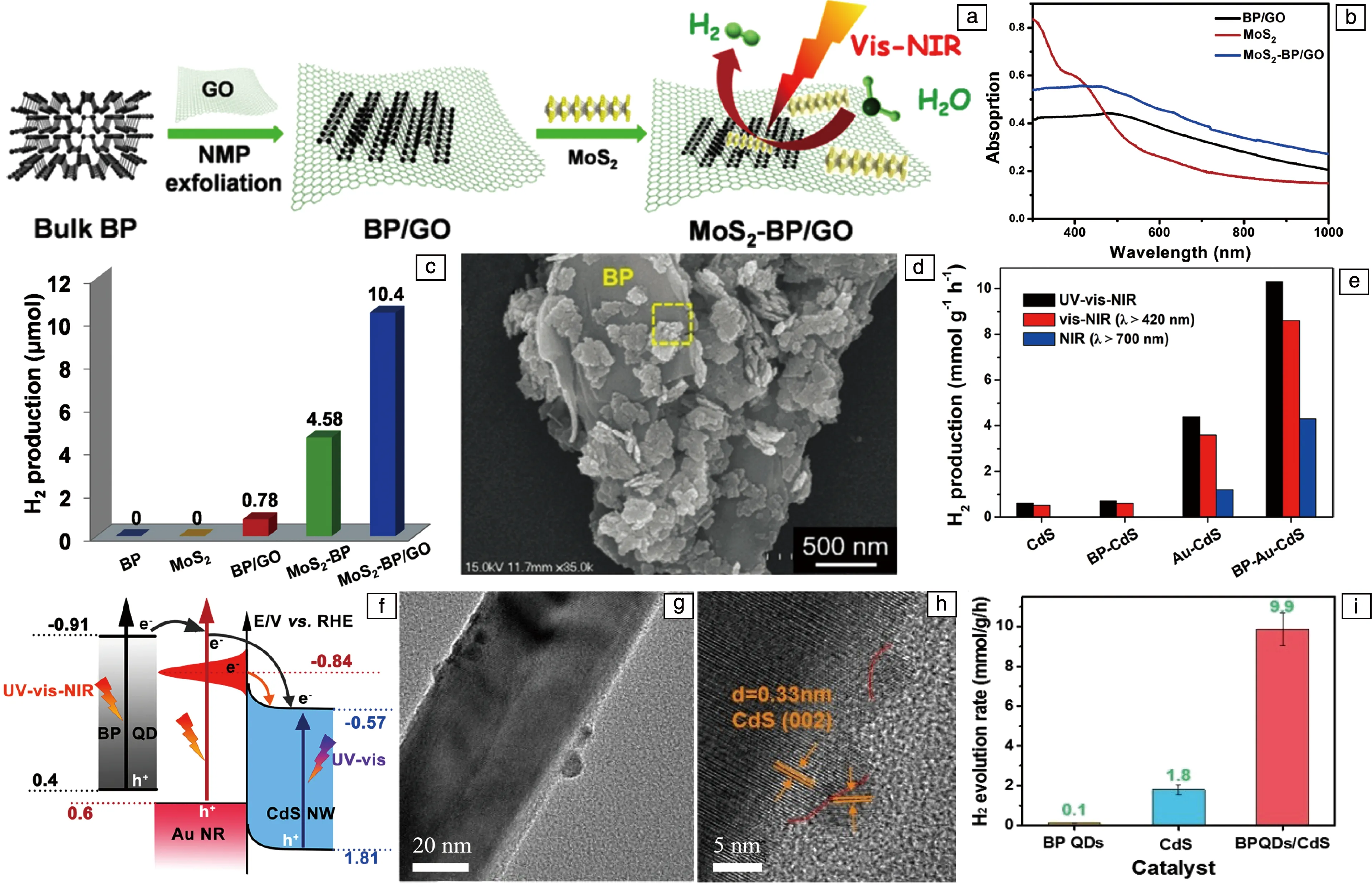
图4 MoS2-BP/GO材料的制备过程示意图(a),UV-vis光谱(b),光催化性能(c)[70];BP/MBWO材料的TEM照片(d)[73];BP-Au-CdS材料的光催化产氢性能(e)和电子转移过程(f)[75];BPQDs/CdS的TEM照片(g),HRTEM照片(h)和可见光下光催化性能(i)[76]Fig.4 Scheme of synthetic process of MoS2-BP/GO (a); UV-vis absorption spectra of as-prepared BP/GO, MoS2, and MoS2-BP/GO (b); photocatalytic H2 production with different catalysts under visible-NIR light irradiation for 3 h (c)[70]; TEM images of 12% BP/MBWO heterojunction (d)[73]; comparison of H2 production rate over different samples under UV-vis-NIR, vis-NIR, and NIR light irradiation (e); schematic illustration of electron-transfer pathway in the BP-Au-CdS system (f)[75]; TEM (g) and HRTEM (h) images of BPQDs/CdS composites; pohotocatalytic H2 evolution rates over various samples under visible light (λ>420 nm) irradiation (i) [76]
Au作为不可再生的贵金属元素,具有储量低、价格高等缺点,因此研究非贵金属催化剂,是十分有意义的。2020年,Liu等将BPQDs与CdS纳米线两种无机非金属材料结合,形成0D-1D异质结用于光催化产氢[76]。通过液相剥离得到BPQDs,将其与水热法合成的CdS纳米线在水溶液中通过静电自组装的方式结合成异质结。TEM照片(图4g)证明BPQDs附着在CdS纳米线的表面形成紧密接触。由HRTEM(图4h)可以发现间距为0.3和0.22 nm的晶格条纹,分别对应于CdS的(002)和黑磷的(002)晶面。在可见光下进行光催化产氢测试(图4i), BPQDs与CdS均展现出较低的催化性能,分别为0.1和1.8 mmol·h-1·g-1,而当BPQDs在CdS表面的负载量为3%时,其催化性能最高,达到9.9 mmol·h-1·g-1。BPQDs与CdS结合后形成Type-Ⅰ型异质结,得益于BPQDs出色的载流子迁移率,CdS被光激发后,其导带电子可以快速转移到BPQDs表面的活性位点,还原水产生H2。CdS价带的空穴迁移到BPQDs的价带后与牺牲剂乳酸反应而消耗,从而实现高效载流子分离,最终达到优秀的光催化产氢性能。
4.2 光催化固氮
2018年,Qiu等将黑磷与g-C3N4结合,使两者之间形成C—P键并用于光催化固氮[77]。通过液相超声法分别剥离得到黑磷纳米片(BPNS)和g-C3N4(CNS)纳米片,以一定的比例混合后再次超声,最后在Ar气下退火即得到BPCNS催化剂。在可见光下进行光催化固氮测试,其结果如图5a所示。纯BPNS、CNS和未退火的0.05BPCNS表现出相似的催化性能,且性能均很低。最高的光催化固氮性能在黑磷含量占0.05时取得,347.5 μmol·L-1·h-1,达到了同等比例的负载Pt催化剂的催化性能,且保持了良好的稳定性。但随着黑磷含量的进一步提升,催化性能反而会出现下降,可能是因为助催化剂的过度负载导致了更多载流子复合。通过XPS分析(图5b和5c)发现,相比于纯黑磷纳米片,BPCNS的P 2p3/2与P 2p1/2峰向更低的结合能方向产生了偏移;与之对应的是,BPCNS的C 1s相比于纯CNS向更高的结合能处发生了偏移。这些偏移可能是由部分电子从富含电子的CNS转移至BPNS导致的。电子转移降低了C原子的外部电子密度,并削弱了对内层电子的屏蔽作用,从而导致C 1s的结合能增加。同时,P原子的电子密度增加,这导致P 2p3/2与P 2p1/2的结合能降低。由此作者认为在0.05BPCNS的合成过程中,C和P之间形成了很强的共价键,引起 C和P原子的电子密度重新分布。通过UV-vis吸收光谱计算不同材料的禁带宽度,结合VBXPS结果,确定两者结合后的异质结结构。BPCNS比CNS有着更负的导带位置,意味着具有更好的还原性能,这是其催化性能更优的一个原因。通过测试0.05BPCNS和CNS的光电流响应(图5d),发现在光照时前者比后者有着显著更高的电流密度。最终确定BPCNS催化性能提升的原因:首先,BP与CNS之间的C—P键加速了电子的迁移,从而提高了光生载流子的分离效率;其次,C—P键的存在使得C与P原子周围的电子重新分布,P原子是电子供体,也是活性位点。
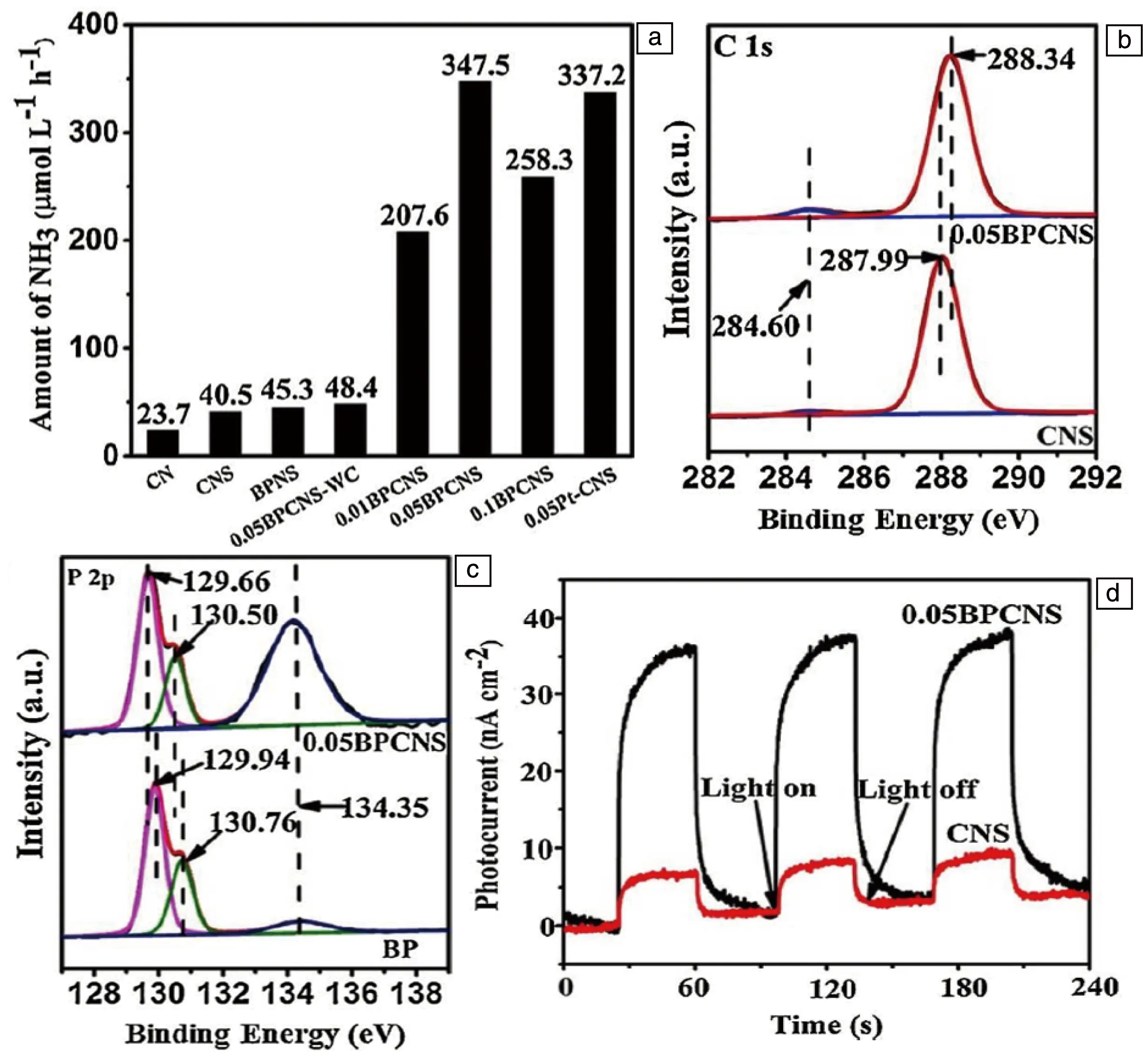
图5 不同材料的光催化固氮性能(a);0.05BPCNS、CNS、BP的XPS图谱:C 1s (b), P 2p (c); 0.05BPCNS与CNS的光电流响应(d)[77]Fig.5 Average reaction rate of photocatalytic nitrogen fixation on CN, CNS, BPNS, 0.05BPCNS-WC and BPCNS (a), XPS spectra of C 1s in CNS and 0.05BPCNS (b), XPS spectra of P 2p in BP and 0.05BPCNS(c), transient photocurrent response for CNS and 0.05BPCNS (d)[77]
2020年,Yu等通过化学刻蚀法开发了具有多边界特征的多晶黑磷纳米片(eBP NFs)[78]。将黑磷晶体与2-溴乙基苯[(2-bromoethyl)-benzene]分散在NMP溶液中,加入一定量的KOH和去离子水,在100 ℃下搅拌加热3 h,通过13 000 r/min高速离心10 min,所得固体用水、乙醇、DMF、NMP清洗后溶于水中,再经过7000 r/min离心10 min即可得到具有多边界特征的黑磷纳米片。电镜表征如图6a~6d所示,由TEM照片可知,所得纳米片的平均尺寸在90 nm左右,且呈厚度不均匀状态。由AFM照片得到纳米片的平均厚度为10 nm左右,表面随机覆盖3 nm的颗粒。由此确定所得到的黑磷纳米片表面崎岖不平,边缘和角落很多。由HRTEM照片发现单个黑磷纳米片中有多个单独的晶体区域,不同区域内的晶格条纹属于不同的黑磷晶面。每个eBP NF都由许多单个细小的黑磷晶粒构成,黑磷晶粒的随机堆积使得eBP NFs厚度不均匀以及表面粗糙,从而在表面边缘和拐角处存在许多晶界,形成独特的“多晶”结构。在以0.05 mol/L的Na2S/Na2SO3为牺牲剂的情况下,对eBP NFs进行光催化固氮测试(图6e)。具有多边界特征的eBP NFs的催化性能为2.37 μmol·L-1·h-1,且8 h的稳定性测试后其催化性能没有明显下降。作为对比,对普通黑磷纳米片(BP NFs)在相同的条件下经行测试,催化性能仅有0.9 μmol·L-1·h-1,远低于eBP NFs。由此证明黑磷纳米片的边缘在光催化氮还原的过程中具有重要作用。光催化固氮机制和催化示意图如图6f和6g所示,N2分子吸附在eBP NFs的边缘,黑磷被光激发后产生光生电子-空穴对,电子在黑磷的导带将N2还原为氨。eBP NFs出色的光催化性能可以归因于其独特的结构和丰富的边缘,因此可以在没有助催化的情况下,表现出不错的催化性能。
2020年,Yuan等通过将黑磷与CdS复合形成非金属异质结催化剂(BPNS/CdS),用于光催化氮还原[79]。首先通过电化学膨胀法制备得到黑磷纳米片(BPNSs),将其分散在乙二醇溶液中,加入一定量的醋酸镉和硫脲(Thiourea),在N2气氛中160 ℃加热16 h,从而在黑磷纳米片表面直接生长CdS。通过TEM(图6h)观察两者结合后的状态,可以发现一个层状结构被许多小黑点覆盖,即CdS均匀地生长在黑磷纳米片的表面,且没有发生团聚。另外,与纯CdS相比,其形貌及尺寸没有改变,CdS颗粒的尺寸在50 nm左右。从照片中还可以发现,CdS几乎完全覆盖了黑磷纳米片,这种现象一定程度上可以阻碍O2与黑磷的接触,减缓黑磷的降解而提高稳定性。UV-vis光谱测试发现黑磷纳米片在500~800 nm可见光区域表现出强吸收特性。相比于纯CdS材料,BPNS/CdS异质结材料在可见光区的吸收性能随黑磷含量的提升而增强。在以甲醇为牺牲剂的情况下,对材料在可见光(λ>420 nm)下进行6 h的光催化氮还原测试,性能结果如图6i所示。受限于低的载流子分离效率,纯CdS表现出最低光催化氮还原性能,随着黑磷含量的提升,催化性能逐渐上升,并在含量为1.5%时达到最大值,为57.64 μmol·L-1·h-1,是纯CdS的3.69倍。这种变化趋势的原因可能是,黑磷含量的提升引入了更多的活性位点,从而性能提高。而当黑磷含量过多时,两者之间的互相遮蔽影响了活性位点的暴露,降低了催化性能。通过理论计算研究异质结界面之间的情况,如图6j~6l所示。在异质结界面处存在明显的电荷重新分配,由于六方CdS的费米能级位置(-0.06 Vvs. RHE)比黑磷的(0.46 Vvs. RHE)更负,因而黑磷可以从CdS上获得电子。另外,异质结界面处的势垒低于费米能级,意味着电子在此处无障碍迁移。理论计算证明了BPNS/CdS异质结可以加速载流子迁移,降低电子空穴对的复合几率,提高光催化固氮性能。最终得到BPNS/CdS的光催化固氮机制,CdS导带上的光生电子迁移到黑磷的导带,在此处发生氮还原反应。黑磷的边缘是反应的活性位点。
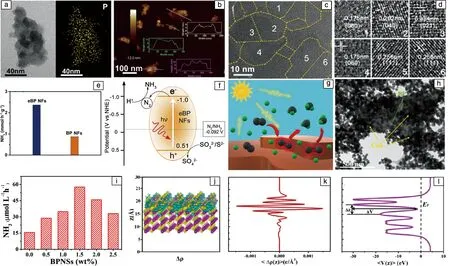
图6 多晶黑磷纳米片(eBP NF)的HRTEM照片和对应的EDS元素分布图谱(a),AFM照片(b)和HRTEM照片(c),对应于图6c中区域的晶格图像(d);eBP NF和BP NFs的光催化氮还原性能(e),eBP NF可能的催化机制(f,g)[78]; 1.5%BPNS/CdS的TEM照片(h),BPNS/CdS的光催化氮还原性能图像(i),BPNS/CdS异质结处理论计算(j-l)[79]Fig.6 TEM image and EDS map of an individual eBP NFs (a); AFM image of an individual eBP NFs(b); HRTEM image of eBP NFs(c); lattice fringes of different areas shown in fig.6c (d); photocatalytic ammonia production rates using eBP NFs and BP NFs as the catalyst (e); schematic mechanism of semiconductor-based photocatalysis for ammonia fixation (f, g)[78]; SEM image of 1.5% BPNS/CdS photocatalyst (h); NH3 evolution rates of various samples under visible light irradiation (λ>420 nm) (i); theory calculation of BPNS/CdS heterojunction (j-l)[79]
4.3 光催化CO2还原
2020年,Zhong等将黑磷与共价三嗪骨架(covalent triazine framework,CTF)合成非金属2D/2D的CTF-BP材料[80]。将二维CTF加入到含黑磷的NMP/乙醇混合液,通过超声与搅拌使两者自组装形成CTF-BP异质结。XPS测试发现,CTF-BP中P 2p的3个峰相比于纯黑磷材料向更低结合能出现了偏移,而在N 1s图谱中,CTF-BP的峰相比于CTF向更高能级处发生了偏移。在Raman图谱中出现了相似的现象,由此证明CTF与BP之间存在异质结交互作用。经过10 h的光催化CO2还原测试(图7a),CTF-BP的CO产量达到46.0 μmol·g-1(4.53 μmol·g-1·h-1),分别是纯黑磷与CTF的3倍与2倍,CH4的产量为78.1 μmol·g-1(7.68 μmol·g-1·h-1),分别是纯黑磷与CTF的23倍与16倍,且没有氢气产生。证明CTF-BP对于催化CO2还原成CO与CH4具有良好的性能。通过理论计算分析其催化机理,从动力学角度看,CTF-BP上的光生电子与CO2结合形成CO2·-,经过两电子反应,形成CO*,且CO*与CTTF-BP之间存在强烈的相互作用,从而增强了8电子反应将CO2还原为CH4。在整个催化过程中,电子由CTF迁移到黑磷上,黑磷是催化反应的活性位点。CTF与黑磷结合后,促进了磷原子与CO*的结合,从而提高了生成CH4的选择性。此外,增强的还原电位、集中的电子密度、电荷转移的协同作用都有利于提高其催化选择性。

图7 BP、CTF、CTF-BP经过12 h的光催化CO2还原性能(a)[80];CsPbBr3/BP的TEM(b)和HRTEM(c)照片;不同材料经过3 h的光催化CO2还原性能(d); CsPbBr3和BP的能带图(e)[81];BW-3的TEM(f)和HRTEM(g)照片;不同材料的光催化CO2还原性能(前1 h),BW-1、BW-2、BW-3、BW-4分别代表黑磷的含量为1%,3%,7%,10%(h)[82]Fig.7 Averaged production rates of CO and CH4 by BP, CTF, CTF-BP (a)[80]; TEM (b) and HRTEM (c) images of CsPbBr3/BP; effect of weight ratio of BP in CsPbBr3/BP for photocatalytic CO2 reduction to CH4 (blue) and CO (red) after 3 h (d); scheme of the band diagrams of CsPbBr3 and BP versus vacuum and NHE at pH=0 (e)[81]; TEM (f) and HRTEM (g) images of BW-3; comparison of photocatalytic activity of the first hour (BW-1, BW-2, BW-3, BW-4 represent the content of black phosphorus is 1%, 3%, 7%, and 10% respectively) (h)[82]
2020年,Wang等通过液相超声法制备黑磷纳米片后,在其表面负载CsPbBr3钙钛矿纳米晶,形成CsPbBr3/BP光催化剂[81]。TEM照片(图7b和7c)显示,小尺寸的CsPbBr3颗粒分布在黑磷纳米片表面,由相应的HRTEM证明,间距为0.41 nm的晶格条纹属于CsPbBr3纳米晶的(110)晶面,间距为0.26 nm的晶格条纹属于黑磷的(040)晶面。经过3 h的光催化CO2还原测试,不同材料的还原性能如图7d所示。黑磷含量为5%时,CsPbBr3/BP材料表现出最好的性能,CH4和CO的产量分别达到32 μmol·g-1(10.7 μmol·g-1·h-1)和134 μmol·g-1(44.7 μmol·g-1·h-1),均高于纯黑磷和CsPbBr3的性能。通过与其他报道的催化剂的催化性能比较,作者认为将黑磷应用于光催化CO2还原是具有意义的。理论计算研究表明两者界面之间存在高的电荷密度和差分电荷密度,即CsPbBr3与黑磷之间形成共价键,从而引起电子的定向迁移,由CsPbBr3转移到了黑磷上。通过热力学研究CsPbBr3与CsPbBr3/BP的光催化反应路径,CO2气体在催化剂表面吸附并活化后进行质子耦合电子转移以形成COOH*,为催化反应的热力学限制步骤,而这一过程,CsPbBr3/BP的自由能势垒低于纯CsPbBr3,即黑磷的引入降低了反应的能垒。最终得到反应的机理如图7e所示,CsPbBr3在被光激发产生光生电子后,电子转移到黑磷上发生CO2还原反应,黑磷是催化反应的活性中心。
2020年,Gao等通过液相超声法制备黑磷量子点,使用水热法合成WO3纳米线,将两者组成0D-1D的BPQD-WO3异质结催化剂[82]。由TEM照片(图7f和7g)可知,BPQDs在WO3表面有着良好的负载,且没有发生团聚,通过HRTEM确认了纳米线是WO3,表面颗粒是黑磷。实验表明,BPQD-WO3异质结材料在连续光照射下表现出独特的光催化活性,如图7h所示。在第1 h内,BW-2表现出最高的CO生成速率72.47 μmol·g-1,而BW-3表现出最高的C2H4生成速率5.92 μmol·g-1。这意味着随着黑磷负载量的提升,更多的CO分子能够偶联在一起,进行连续的氢化过程以生成C2H4。但是当黑磷的含量进一步提升后,CO和C2H4的产量均出现明显下降,可能是因为WO3表面活性位点被黑磷量子点占据,对催化反应产生了抑制。两者形成异质结后的催化性能显著高于纯BPQD。
C2H4作为重要的工业原料,其生产具有非常重要的意义,在以往报道的基于WO3的催化剂中,均没有检测到C2H4的生成,但是在BPQD-BiVO4材料中发现了催化CO2还原为C2H4的性能,故作者推断BPQD在此过程中起到了关键作用。通过理论计算对黑磷的催化过程进行研究,分别对黑磷Armchair边缘和Zigzag边缘的反应热力学进行计算。结果表明,在AC边缘上,速率限制步骤是第一个电子-空穴对转移形成*CO—COH的过程,其能垒为-0.3 eV。在ZZ边缘上,速率限制步骤是*CO—COH氢化为*CO—CHOH的过程,其能垒为-0.7 eV。因此在AC边缘上更易于发生CO2还原成C2H4的反应,且能垒比报道的其他材料均低。此外,BPQDs使得C—C的二聚化更易于发生,将催化剂表面形成CO的可能性降到最低。BPQD-WO3材料形成Z-型异质结后,在光激发下,可以发生有效的空间电荷分离,WO3导带上的电子与黑磷价带上的空穴复合,增强黑磷导带上的CO2还原反应,同时也降低了自氧化的趋势,提高了稳定性。
4.4 其他光催化应用
2020年,Wang等将黑磷量子点与管状g-C3N4(TCN)结合,形成0D-1D异质结催化剂(BPTCN),用于去除各种有机污染物(盐酸土霉素(OTC-HCl),四环素(TC)、罗丹明B)和重金属Cr(VI)离子[83]。由SEM和TEM观察可知得到的BPQDs的平均尺寸为3.32 nm,TCN的直径在6~8 μm。通过TEM与EDS元素分布分析证明BPQDs均匀附着在TCN表面。在OTC-HCl的降解的测试(图8a)中,首先将催化剂和OTC-HCl在避光条件下搅拌2 h,几乎没有OTC-HCl被降解。将其转移到可见光下照射1 h,BPTCN对OTC-HCl的降解率达到了81.05%,表观速率常数(Kapp)为0.0276 min-1,是TCN的2.36倍。这可以归因于TCN表面负载的BPQDs提高了载流子分离效率,减少了电子-空穴对的复合。由此证明,两者之间的紧密结合有利于提升OTC-HCl降解速率。此外,实验表明BPTCN在可见光下对四环素和罗丹明B的降解速率分别高达77.11%和99.43%。证明该材料具有优异的有机污染物降解性能。
为了进一步研究BPTCN的催化能力,还将其用于水溶液中Cr(VI)的热还原(图8b)。BPTCN表现出最佳的Cr(VI)去除能力(94.71%),而BPQD、CN和TCN分别实现了17.61%,22.49%和81.59%的Cr (VI)光还原。BPTCN显著提高的光还原能力可能归因于在管状g-C3N4表面负载BPQD引起的空间电荷的快速分离和转移。从溶液的颜色变化也能直观地证明Cr(VI)的浓度降低。通过相关测试和理论计算,得到BPTCN催化性能提升的原因:一方面,一维多孔管状结构减少了载流子到表面的扩散距离;另一方面,BPQDs被均匀地负载在TCN的表面上,获得独特的0D-1D结构,形成了界面P—C键,共同阻碍了载流子的复合;此外,BPQDs的加入极大地提高了BPTCN的光吸收性能。由此证明BPQDs对于BPTCN催化性能的提升起到了关键作用。
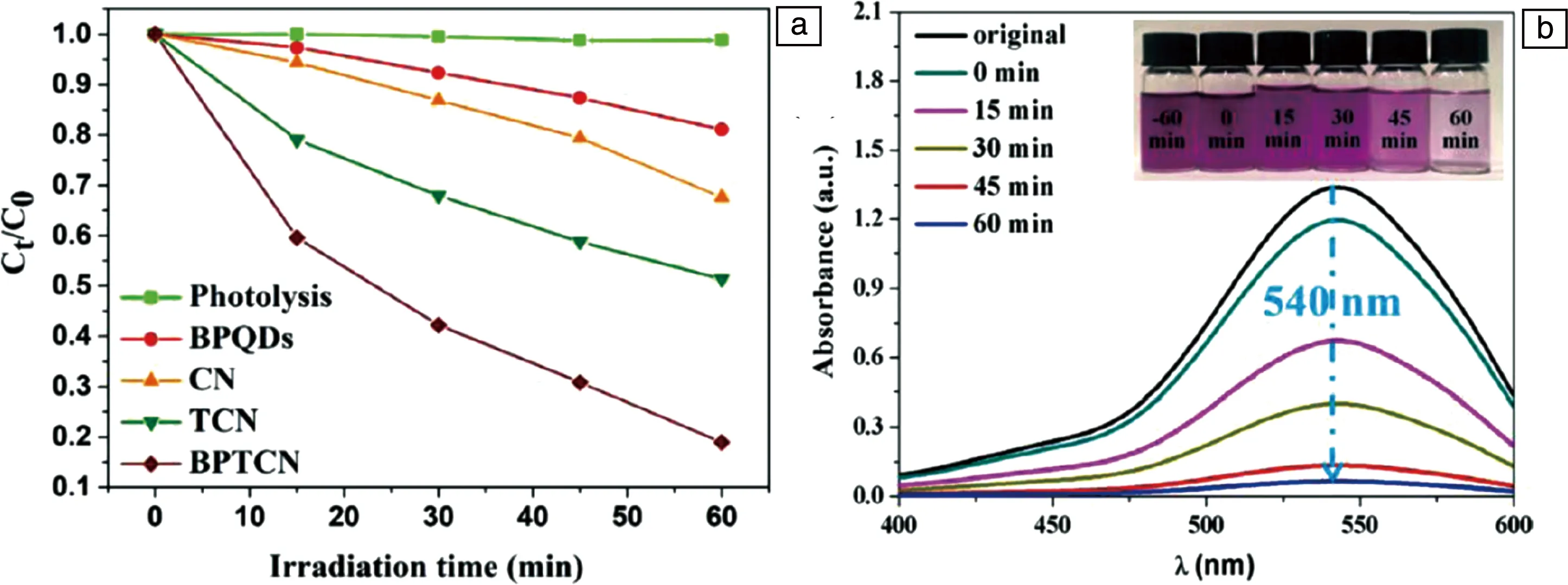
图8 光催化降解OTC-HCl性能(a),Cr (VI)的热还原性能(插入图为催化不同时间后溶液的颜色照片)(b)[83]Fig.8 Photocatalytic degradation of OTC-HCl over CN, TCN, and BPTCN (a), temporal absorption spectra of Cr (VI) with BPTCN catalysts under visible light irradiation (the inset in Fig.8b is the photographs of Cr (VI) aqueous solution after reaction) (b) [83]
4.5 小结
本章详细介绍了黑磷在光催化领域的应用,包括光催化产氢、光催化固氮、光催化CO2还原和有机物降解等方面,不同应用的性能对比如表2所示。在光催化应用上,黑磷展现出诸多独特的优点:如丰富的形貌,大量的活性位点、广泛的光吸收范围等,以及良好的与其他材料组合适应性,使得黑磷在光催化中能发挥多重作用。除了上述优点,黑磷也表现出一些问题,比如其易于氧化的特点,对于光催化的循环稳定性有非常大的影响,使得其对反应环境比较苛刻。此外,黑磷纳米片及量子点较低的产量是阻碍黑磷快速应用与更广泛研究的主要原因。最后,人们对黑磷在光催化过程中的作用机理已有一定的了解,但依然需要更深层次的研究,从而更有针对性地对黑磷材料进行优化,提高其催化性能和稳定性。
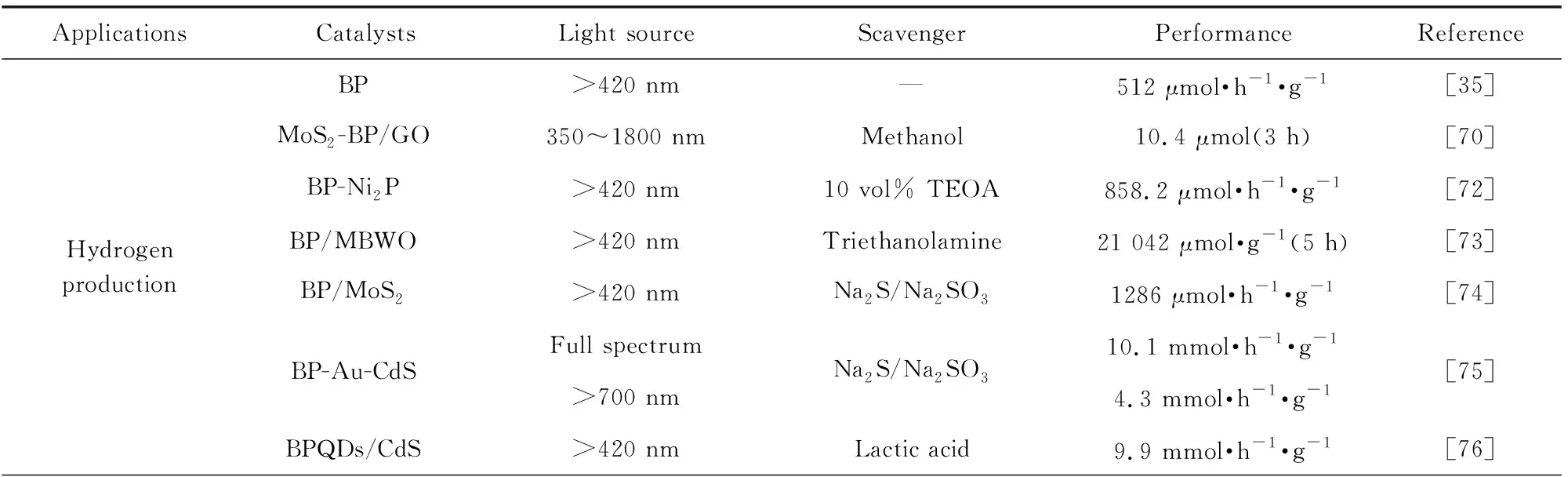
表2 黑磷催化剂在不同光催化应用的性能对比
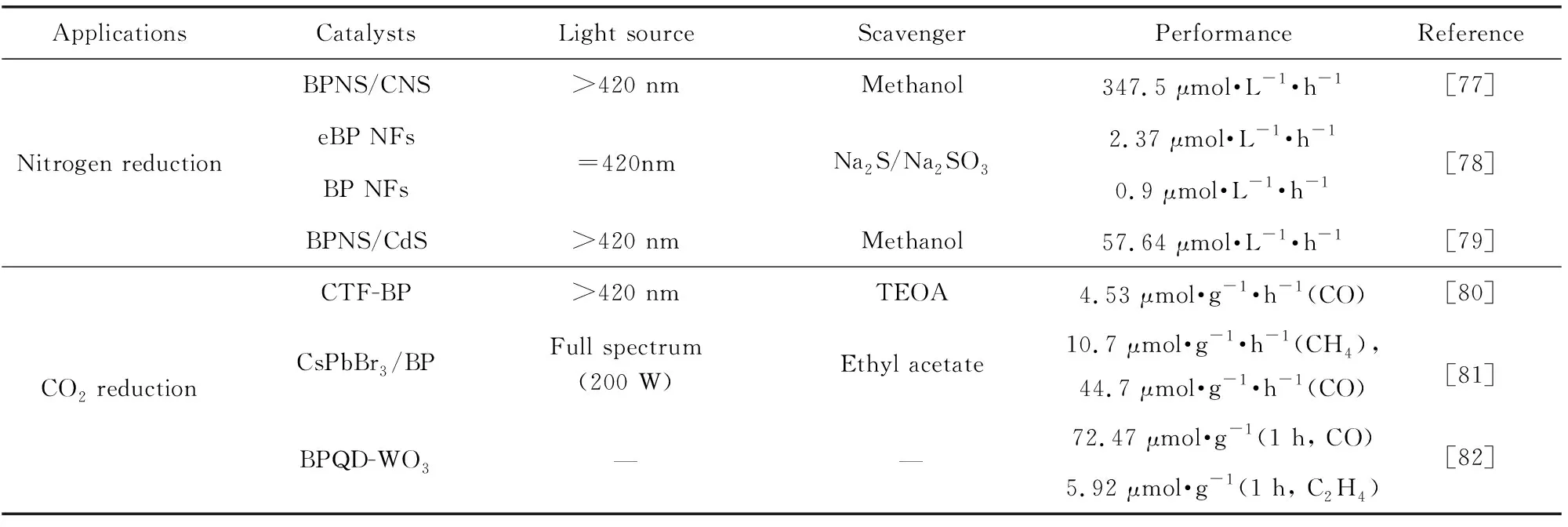
续表
5 结 语
在能源危机和环境保护问题的多重压力下,全面推进新能源开发与利用、工业生产绿色化和污染治理已经成为未来工作的重点。光催化技术以太阳光作为能量来源,不仅获取方便、绿色清洁,而且还具有可持续发展性,相应的光催化产氢、固氮及CO2还原技术发展迅猛,可以生产清洁能源、减少污染物排放,极具研究与应用意义。
黑磷材料在光催化领域表现出优异的性能,已经广泛应用于光催化产氢、固氮和CO2还原等应用,其原因如下:① 黑磷材料具有紫外-可见-近红外广阔的光吸收范围,有利于提高催化材料的光吸收效率;② 黑磷材料具有优异的载流子迁移率,可提高载流子分离效率;③ 黑磷具有二维结构,具有独特的表/界面特性;④ 黑磷纳米片具有极大的比表面积,具有大量的活性位点。
虽然目前黑磷材料在光催化领域已经展现了一定的应用前景,但依然需要更深入的研究。比如,目前黑磷在光催化固氮上的应用依然偏少,有待研究人员进一步探索。此外,黑磷材料表面丰富的活性位点,一方面有利于促进催化反应,另一方面也使其易于与氧气反应而降解,因此其制备、合成以及保存均需要在特殊条件下进行,提高了应用成本。探究有效的抑制黑磷降解的措施,同时最大限度地保留黑磷的催化活性,是尤为关键的一点。最后,目前对黑磷材料在催化过程中的作用及原理尚不明确,需要研究人员从实验表征以及理论模拟计算等多个角度进行深入探究,只有在清楚了解催化过程及机理的基础上,才能针对性地优化材料,有效提高黑磷的催化效率。
本文对黑磷的结构、黑磷的特性、黑磷材料的制备方法进行简单的介绍,结合黑磷材料在光催化领域中的实例研究,总结了黑磷材料在光催化方面的应用,讨论了黑磷材料在光催化中的作用。黑磷作为一种独特的新型材料,已经展现出广阔的应用前景,但对于其发展,依然需要研究人员进一步的深入探索。
猜你喜欢
无机盐工业(2021年6期)2021-06-23
物理化学学报(2020年7期)2020-07-23
磷肥与复肥(2020年2期)2020-05-06
无机盐工业(2017年5期)2017-05-25
郑州大学学报(理学版)(2017年1期)2017-04-07
化工管理(2017年25期)2017-03-05
贵州师范学院学报(2016年3期)2016-12-01
云南师范大学学报(自然科学版)(2015年5期)2015-12-26
中央民族大学学报(自然科学版)(2015年2期)2015-06-09
物理实验(2015年10期)2015-02-28
