
稳定同位素稀释-液相色谱串联质谱法测定血清中的25-羟基维生素D2和25-羟基维生素D3含量
2021-12-08 00:33刘艳琴彭莹莹刘圆圆赵少志张欣文
分析仪器 2021年6期
刘艳琴 张 璇 彭莹莹 刘圆圆 赵少志 张欣文
(西安市人民医院(西安市第四医院),西安 710000)
1 前言
维生素D是人体一种重要的脂溶性维生素,多项研究表明,充足的维生素D含量不仅对维持人体骨骼发育、调节钙磷水平有重要作用,同时,对加强免疫反应、抑制抗炎反应,提升肝脏抗氧化能力、调控血糖水平及抑制癌症发生等均有积极作用[1,2]。维生素D在体内主要有维生素D2、维生素D3两种存在形式,其中维生素D2主要通过植物性食物摄取得到补充,维生素D3主要通过动物性食物摄取和紫外照射合成,此两种维生素体内没有生理活性,经循环、转化后形成活化状态的1,25-二羟维生素D3/D2(1,25(OH)-D3/D2)、24,25-二羟维生素D3(24,25(OH)-D3/D2)、25-羟基维生素D2/D2(25(OH)-D2/D2)和25-羟基维生素D3/D2(25(OH)-D3/D2)等形式,而由于25(OH)-D3/D2在体内含量高、半衰期长、不易分解,因而成为检测体内维生素D含量的最佳指标[3,4]。
维生素D缺乏或过量均会引发多种疾病,维生素D缺乏时,会引发儿童成长缓慢、免疫学疾病、代谢性疾病,成人细菌性感染、心脑血管疾病等,孕妇妊娠糖尿病、产后骨质疏松等,而过量维生素D的摄入会引发肌无力、骨痛、高钙血症等疾病,严重影响人类的身体健康[5,6]。
据统计分析,全球约一半人口存在维生素D缺乏情况,且各国人口中维生素D缺乏率各不相同,而造成这一统计差异的一个重要原因是各国检测维生素D含量方法的差异,因此,建立一种高效、精准的检测方法对评估人体内维生素D含量存在重要意义[7]。目前,体内维生素D的检测方法有电化学发光免疫检测法(ECLI)[8]、放射免疫法[9]、酶联免疫法(ELISA)[10]、液相色谱(HPLC)法和液相色谱-串联质谱(HPLC-MS/MS)法[11,12],而免疫法具有无法同时检测样本中25(OH)-D3/D2含量、存在抗体反应交叉反应,易影响实验精准度等缺点[13]。随着科研的发展,HPLC-MS/MS法已经被认为是检测人体25(OH)-D2和25(OH)-D3的金标准,而由于各实验室采用HPLC-MS/MS法检测人体25(OH)-D2和25(OH)-D3方法不尽相同,如郑仁锦等人通过乙腈、饱和硫酸锌、正己烷提取、沉淀、萃取等步骤处理样本后质谱检测,进样量为15μL,检测时间10min,于伟越等人通过甲醇、正己烷、硫酸锌提取、萃取、沉淀等步骤处理样本后质谱检测,进样量为25μL,检测时间10min[14-16]。同时,为了方便处理样本,多数机构因血斑不受地域限制、运输便捷、利于存储等优势以血斑为样本检测,但血斑样本制作过程繁琐、前处理步骤较多,人工操作过程中易出现误差,影响实验结果[17,18]。基于以上原因,本方法通过直接处理血清样本进行试验,优化实验条件,简化实验流程,缩短检测时间,添加了衍生剂,增强信号强度,建立了一种操作简单、快速高效、精准、高灵敏度的检测血清样本中25(OH)-D2和25(OH)-D3的HPLC-MS/MS方法。
2 实验和方法
2.1 仪器和试剂
2.1.1仪器
液相色谱-串联质谱(Waters Xevo TQD三重四极杆液质联用仪);C18色谱柱(Kinetex,30×2.1mm,粒径1.3μm);DC150-1A 96孔氮吹仪;微孔板恒温振荡器(型号:MB100-4A),96孔深孔样品板(2ML/孔);检验分析用纯水设备(型号:TCHS-05 RO/2OF)。
25-羟基维生素 D2标准品(纯度95%);25-羟基维生素 D3标准品(纯度98%);25-羟基维生素D2-氘代同位素内标(25(OH)-D2-IS)、25-羟基维生素D3-氘代同位素内标(25(OH)-D3-IS)(纯度>98%);4-苯基-1,2,4-三唑啉-3,5-二酮(PTAD)、甲醇、甲酸、乙酸乙酯(HPLC级试剂)。
2.1.2试剂的配制
将干燥后的内标物质溶于1100μL甲醇,配置成内标储备液,分装后-20℃保存,使用时按照实验配置所需浓度的提取液。将PTAD溶于乙酸乙酯,配置后PTAD浓度为0.2μg/μL。
2.1.3液相色谱-串联质谱条件
ESI离子源温度为150℃,脱溶剂气温度为500℃,脱溶剂气流量为800L/hr,流动相:A相(甲酸∶乙酸铵水,v/v/v=1/1/1000),B相(甲酸∶甲醇,v/v=1/1000),以0.4mL/min流速,进样器温度10 ℃,柱温40℃,上样量10μL,0~1.1min、1.1~1.7min、1.8~3.5min以40%B、98%B、98%B流动相梯度洗脱样本,以40%B冲洗管路。各分析物质谱扫描参数见表1。

表1 各分析物MS扫描参数
2.2 标准品和样本的制备
抽取静脉血3mL于采血管中,以3500r/min转速离心5min血样,取上清备用(如当天实验,样本存于4℃,如隔天实验,冻存于-20℃,实验前及时拿出解冻),分别各取100μL上清样本、标准品、25(OH)-D2和25(OH)-D3的低质控品(浓度分别为3ng/mL和15ng/mL),25(OH)-D2和25(OH)-D3的高质控品(浓度分别为15ng/mL和75ng/mL)于1mL96孔板中,加400μL提取液震荡均匀后,各吸取300μL混合液于96孔板,氮气吹干仪吹干;每个孔中加100μLPTAD衍生化溶液,300rpm、30℃孵育30min;各孔板加50μL纯水作为淬灭剂终止反应,300rpm、30℃孵育5min;静置5min后置于液相色谱-串联质谱中检测。
2.3 标准曲线和定量检出限实验
根据试剂盒提供的系列浓度工作溶液,即分别将25(OH)-D2和25(OH)-D3标准品加400μL内标提取液稀释,25(OH)-D2标准品浓度梯度为0.4ng/mL、0.8ng/mL、2ng/mL、4ng/mL、10ng/mL、20ng/mL,25(OH)-D3标准品浓度梯度为2ng/mL、4ng/mL、10ng/mL、20ng/mL、50ng/mL、100ng/mL,采用血清加标方式配制标准曲线,校准标样和质控样品中的分析物浓度将以分析物与内标的色谱峰面积比为纵坐标,用加权(W=1/X2)最小二乘法以分析物的浓度(X)与峰面积比(Y)进行线性回归分析。
2.4 精密度、准确度和回收率
25(OH)-D2和25(OH)-D3的低质控浓度分别为3ng/mL和15ng/mL,高质控浓度分别为15ng/mL和75ng/mL,将25(OH)-D2和25(OH)-D3的低质控和高质控血清平行制备6份,每份样本按照2.2制备过程制备、2.1.3 LC-MS/MS仪器条件下,每天测定6次,连续测定3天,测定精密度与准确度。
2.5 样品稳定性
血清样品室温稳定性实验:取1份混合血清分别置于室温(10~30℃)0h、2h、4h、8 h,按照2.3制备过程制备、2.2 LC-MS/MS仪器条件下,以0h检测数据为参照,计算其他样本指标成分的相对偏差(RE),以评价血清样品室温稳定性;血清样品冻融稳定性实验:取一份混合血清分别置于<-65℃冰箱,进行0、1、3、6次冻融,按照2.2制备过程制备、2.1.3 LC-MS/MS仪器条件下,以第0 次冻融(未冻融的血清样本)检测数据为参照,计算其他样本指标成分的相对偏差,以评价血清样品冻融稳定性。
2.6 空白加标回收率
空白样本基质中25(OH)-D2和25(OH)-D3检测值分别为0.15±0.13ng/mL和6.96±0.41ng/mL,以低质控和高质控品作为样本加入基质中,操作过程同2.2,每个浓度平行制备6份,平行3组,计算加标回收率。
2.7 残留评价
分别检测25(OH)-D2、25(OH)-D3标准品的最高浓度20ng/mL和100ng/mL后,测试两个空白基质样品,通过测定空白基质样品中的分析物和内标的峰面积,并与最低浓度点校正标样比较,以确认残留对分析物准确定量的影响。
2.8 基质效应评价
以1∶1的体积比在血清中加入低质控品、高质控品溶液,通过比较血清/质控品溶液中分析物峰面积/内标峰面积比值与1/2血清中分析物峰面积/内标峰面积比值和质控品溶液中分析物峰面积/内标峰面积比值评估内标归一化基质因子,以达到评估基质效应的目的。
3 结果与讨论
3.1 条件优化
离子对条件优化:为提高灵敏度,本实验在样本提取时加入了PTAD进行衍生化,通过引入氮原子基团,增加ESI离子源离子化分析物时形成加[M+H]+峰,而由于衍生时,25(OH)-D易发生解离,形成[M-H2O+H]峰,因此,在LC-MS/MS定量分析时,选择[M-H2O+H]+作为母离子。流动相条件优化:PTAD与25(OH)-D2和25(OH)-D3衍生化反应时,会产生两种手性碳原子的化合物,即6R和6S光学异构体构型,为方便定量,定量时选择6S构型为定量分析物,且为增加保留时间,获得良好峰形,本实验在流动相中加入甲酸和乙酸铵,并调节流速和不同时间的流动相比例进行检测。
3.2 方法学验证[19]
3.2.1标准曲线和定量检出限
回归方程(Y=bX+a)即为标准曲线,结果如图1所示。25(OH)-D2和25(OH)-D3的线性相关系数均为R2>0.999,表明线性关系良好;以检测标准曲线线性最低点作为定量检出限考察,即连续进样检测6次,结果显示25(OH)-D2和25(OH)-D3线性最低点的CV和RE均小于20%,表明定量检出限满足方法学评价,见表2。
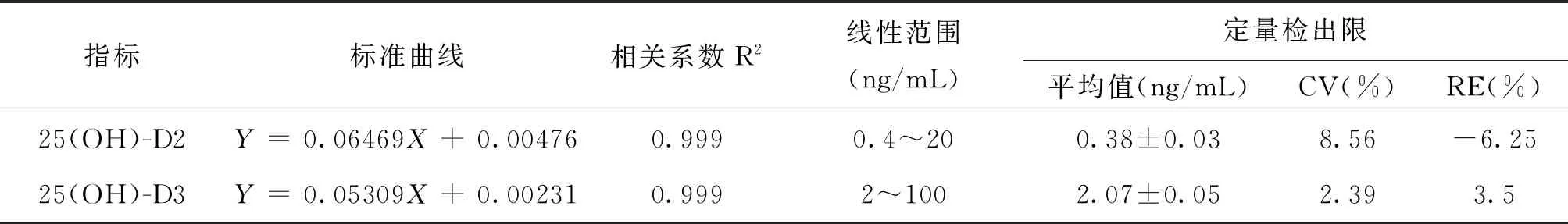
表2 25(OH)-D2和25(OH)-D3的线性范围

图1 标准曲线图与色谱图注:a、c、和b、d分别表示25(OH)-D2和25(OH)-D3的标准曲线图与色谱图
3.2.2精密度、准确度和回收率
测定结果的RSD(%)和RE(%)分别作为考察精密度与准确度的指标,结果如表3所示。25(OH)-D2精密度RSD≤3.55%,准确度-4.67%≤RE≤3.33%,回收率为93.83±8.38%~101.83±5.15%;25(OH)-D3精密度RSD≤6.73%,准确度RE≤2.56%,回收率为97.33±7.28%~100.33±9.09%,各指标精密度、准确度和回收率均良好,满足方法学要求。

表3 25(OH)-D2和25(OH)-D3的精密度和准确度
3.2.3样品稳定性
与0 h放置血清相比,于室温(10~30℃)放置8 h后,血清中25(OH)-D2/D3的RSD≤10.49%,-9.42%≤RE≤0.52%;与无冻融血清相比,冻融6次血清中25-羟基维生素D2/D3的RSD≤7.456%,-3.99%≤RE≤-0.54%,表明血清中25-羟基维生素D2/D3样本稳定性良好(表4)。
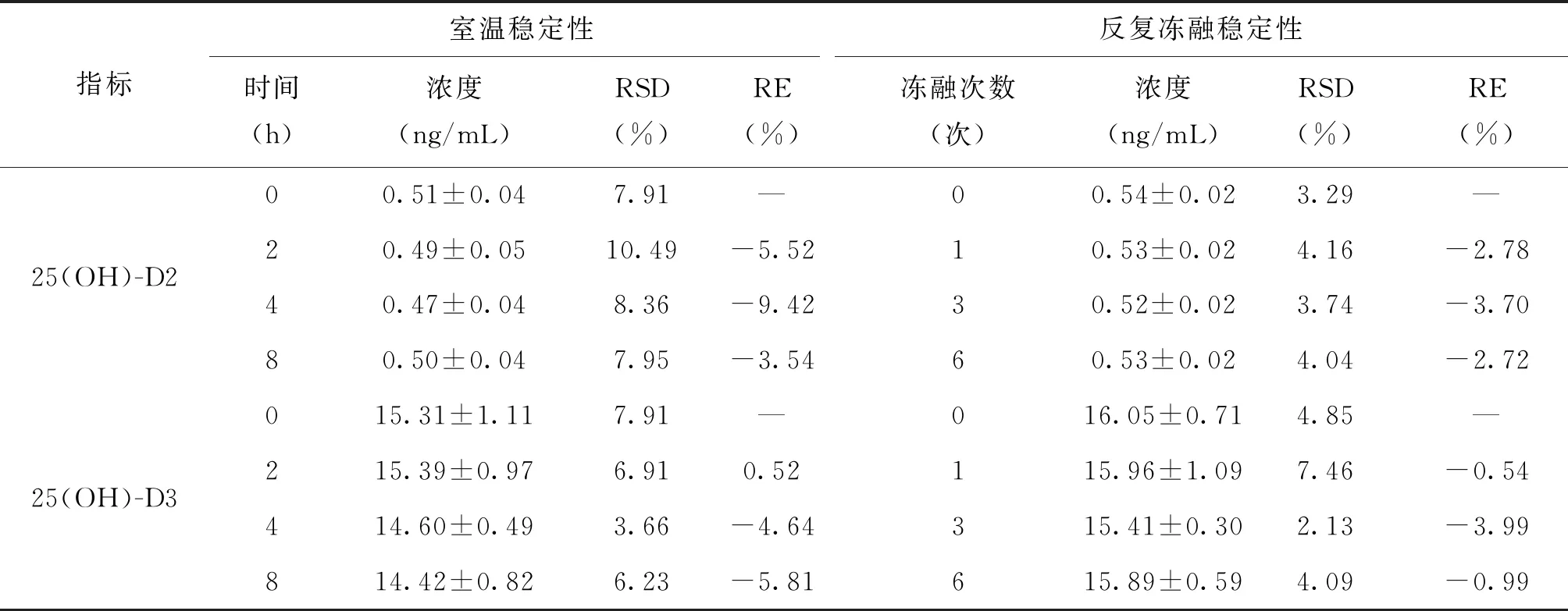
表4 25(OH)-D2和25(OH)-D3的样品稳定性
3.2.4空白加标回收率
如表5所示,25(OH)-D2加标回收率为97.50±9.85%~98.67±9.48%,CV%≤10.1,25(OH)-D3加标回收率为98.33±10.15%~98.5±10.82%,CV%≤10.98,满足验证要求。

表5 25(OH)-D2和25(OH)-D3的加标回收率
3.2.5残留评价
分析物25(OH)D2、25(OH)D3、内标均基本无残留,满足中国药典规定在分析物保留时间处的干扰峰的响应均不超过定量下限中分析物响应的20%,内标保留时间处的干扰峰的响应均低于低浓度样品中内标响应的5%[19],残留实验评价满足方法学评价要求。
3.2.6基质效应评价
有血清或无血清基质中不同加标浓度25(OH)-D2和25(OH)-D3内标归一化基质因子在0.89~0.94之间;即内标与分析物的基质效应接近,可补偿样品中分析物可能出现的基质效应,基质效应不影响最终的准确定量分析,具体结果如表6所示。

表6 25(OH)-D2和25(OH)-D3的基质效应评价
3.3 HPLC-MS/MS法与ELISA方法的比较
为考察HPLC-MS/MS方法与ELISA法测定血清中25(OH)-D2和25(OH)-D3含量的一致性,选取50例正常人群的样本,按照3500r/min转速离心5min后,提取上清,将上清样平均分为两份,一份按照2.3方法制备后,用HPLC-MS/MS方法检测,另外一份按照ELISA试剂盒(欧蒙,批号:E200803AB)操作方法检测;由于ELISA检测无法将25(OH)-D2和25(OH)-D3区分开,因此,HPLC-MS/MS检测结果中将25(OH)-D2和25(OH)-D3含量相加后与ELISA检测值比较相关性。结果如图2所示,x轴、y轴分别为ELISA和HPLC-MS/MS的检测结果,所得线性关系为Y=1.3229X-2.87,相关性系数R2=0.8507,表明两种方法一致性良好。

图2 HPLC-MS/MS和ELISA检测25(OH)-D浓度的线性关系
4 结论
本研究建立了一种通过HPLC-MS/MS法检测25(OH)-D2和25(OH)-D3含量的方法,实验经PTDA衍生化样本,提高了检测的灵敏度,且只需3min即可完成样本的检测,实现了高效、快速、精准检测的目标,同时,经验证,HPLC-MS/MS法与ELISA法检测结果一致性良好,可为不同检验条件下选择合适的检验25(OH)-D2和25(OH)-D3含量的方法,提供科学依据。
猜你喜欢
南昌大学学报(工科版)(2022年2期)2022-07-18
日用电器(2022年3期)2022-04-14
西北药学杂志(2021年3期)2021-12-04
今日农业(2021年9期)2021-11-26
口腔护理用品工业(2021年4期)2021-11-02
中国科技纵横(2019年23期)2019-02-14
食品界(2017年7期)2017-08-24
农村百事通(2017年6期)2017-03-30
海峡科技与产业(2017年1期)2017-03-04
热带农业工程(2014年6期)2015-01-28
