
全基因组重测序揭示青藏高原东缘及东南缘东方蜜蜂遗传多样性与适应性进化
2022-06-09 10:33唐相友宋华莉张小燕唐紫寒王文峰陈新兰周泽扬许金山
昆虫学报 2022年5期
唐相友, 宋华莉, 石 鹏, 张小燕, 唐紫寒,王文峰, 扎 罗, 陈新兰, 周泽扬, 许金山,*
(1. 重庆师范大学生命科学学院, 重庆 401331; 2. 活性物质生物技术教育部工程研究中心,重庆 401331;3. 重庆市媒介昆虫重点实验室,重庆 401331; 4. 西藏农牧学院, 西藏林芝 860000)
青藏高原东缘及东南缘处于青藏高原边缘地带,因其地形复杂、生态境多样化,为各类生物生态、繁衍发展提供了条件,即是全球生物多样性热点地区之一,也是研究物种形成和分化机制的理想区域(王凤英等, 2005; 刘天猛, 2018; 张剑搏等, 2019)。青藏高原东缘及东南缘东方蜜蜂Apiscerana适应了高原复杂多变的环境,其优质种质资源对于蜜蜂遗传改良具有重要意义。因此,开展高原东方蜜蜂遗传多样性及其环境适应性的分子机制研究显得尤为重要。此前,学者主要利用形态学、线粒体DNA和微卫星分子标记的方法探讨了青藏高原东方蜜蜂的遗传多样性,表明在此区域存在阿坝蜜蜂Apisceranaabansis和藏南蜜蜂Apisceranaskorikoui两个不同类型的高原东方蜜蜂遗传群体(杨冠煌等, 1986; Zhuetal., 2017; Yuetal., 2019; 周丹银等, 2019)。
随着我国东方蜜蜂全基因组精细图谱的完成与公开(Wangetal., 2020; Lanetal., 2021),通过全基因组重测序鉴定群体单核苷酸多态性(SNP)变异位点,对特定区域的东方蜜蜂遗传多样性进行更加全面的分析,弥补传统形态学与单一分子标记在东方蜜蜂遗传分化研究中的不足,对挖掘和利用我国蜜蜂遗传资源具有促进作用(Chen NBetal., 2018; Jietal., 2020; Shietal., 2020)。当前,尚缺乏对青藏高原东缘及东南缘东方蜜蜂的遗传多样性的深入研究,区域内不同群体之间的近缘关系及扩散来源也暂未厘清。因此,本研究采集了青藏高原东缘和东南缘及邻近地区的东方蜜蜂样本并进行全基因组重测序,结合此前已公开的部分数据,拟通过群体遗传结构及单倍型分析,研究青藏高原东缘和东南缘东方蜜蜂遗传多样性,并结合受选择分析初步鉴定青藏高原东缘及东南缘东方蜜蜂高原适应性相关基因,初步探讨其潜在的分子适应机制,以期为进一步解析青藏高原东方蜜蜂的遗传资源多样性、种群扩散规律以及适应高原生境下的分子进化机制提供参考。
1 材料与方法
1.1 样本采集与收集
采集青藏高原东缘和东南缘及邻近地区的77群东方蜜蜂,保存至75%的酒精中。从每群中随机抽取1头工蜂,取头、胸部组织提取总DNA,4℃保存待测序;从GenBank数据库下载青藏高原东缘和东南缘及邻近地区的90群东方蜜蜂全基因组重测序原始数据(GenBank登录号: PRJNA418874),上述167群分属于11个地理种群,范围覆盖了青藏高原东缘(邛崃山脉东西、南北延伸以及四川九寨沟)及东南缘(西藏波密以及云南迪庆和西双版纳)区域(表1),以西域黑蜂Apismelliferasinisxinyuan全基因组重测序数据(GenBank登录号: PRJNA301648)作为外群。
1.2 全基因组重测序与SNP calling
委托公司使用Illumina HiSeq测序平台进行对1.1节采集的77群东方蜜蜂进行全基因组重测序(GenBank登录号: PRJNA827659)。使用FASTX-Toolkit软件(http:∥hanno nlab.cshl.edu/fastx_toolk it/)去除低质量(Q<20)序列和含有5%不可读碱基(N碱基)以上的原始reads,获得高质量可用于后续遗传分析的reads。利用比对软件BWA-MEM,将样本reads匹配到高质量东方蜜蜂参考基因组上(Lanetal., 2021),采用bamdst计算样本的匹配率和测序深度。采用SAMtools(Li, 2011)软件对比对BAM文件进行排序处理,而后使用GATK4.0(do Valleetal., 2016)对比对BAM文件去除重复读取和SNP calling,采用HaplotypeCaller和VariantFiltration程序包对SNPs进行鉴定和过滤。采用VCFtools(Daneceketal., 2011)对SNP候选数据集进行进一步筛选,确定SNPs数据用于后续相关分析。
1.3 群体遗传分析
基于高质量SNPs数据,使用软件ADMIXTURE(Alexanderetal., 2009)的基于块松弛算法(block relaxation algorithm)分析群体遗传结构和个体祖先血缘混合情况。 使用EIGENSOFT(Priceetal., 2006)程序进行主成分分析。采用PHYLIP(v3.697)邻接法(neighbor-joining, NJ)基于SNPs构建系统进化树,以西域黑蜂的SNPs作为外群,bootstrap值设置为1 000,使用在线工具iTOL(http:∥itol.embl.de/)进行系统进化树可视化呈现。使用METAPOP2(López-Corteganoetal., 2019)计算种群间最小遗传距离与种群内成对遗传分化指数(Fst)。将青藏高原东缘及东南缘高原东方蜜蜂分别与邻近地区非高原东方蜜蜂组合,即:(1)藏南高原与滇北高原组:波密(BM)+迪庆(DQ)+西双版纳(XSBN);(2)川西高原组:马尔康(MEK)+丹巴(DB)+邛崃山(QLS);(3)川北高原组:九寨沟(JZG)+神农架(SNJ)。利用DnaSP6(Rozasetal., 2017)软件进行线粒体基因组单倍型分析,采用POPART(Leigh and Bryant, 2015)进行图形化展示。
1.4 选择信号分析
为挖掘青藏高原东方蜜蜂环境适应性相关基因,将青藏高原东缘及东南缘高原东方蜜蜂分别与遗传关系较近的非高原东方蜜蜂组合,即:(1)藏南高原组:波密(BM)+西双版纳(XSBN);(2)滇北高原组:迪庆(DQ)+西双版纳(XSBN);(3)川西高原组:丹巴(DB)+邛崃山(QLS);(4)川北高原组:九寨沟(JZG)+神农架(SNJ)。采用VCFTOOLS(Daneceketal., 2011)以滑动窗口大小50 kb,步长10 kb计算核苷酸多态性(θπ)和遗传分化指数(Fst),采用XPCLR(Chenetal., 2010)软件计算XP-CLR值,滑动窗口大小设置为50 kb,步长10 kb,每代碱基突变率设置为5.3e-9。基于东方蜜蜂阿坝株的全基因组序列及注释文件(Lanetal., 2021)提取上述组合中两群体基因组中所有潜在受选择的基因序列,通过Eggnog-mapper(Cantalapiedraetal., 2021)进行GO注释和KEGG通路分析,采用FDR进行校正。
2 结果
2.1 SNP鉴定
与东方蜜蜂阿坝株全基因组精细图谱序列(Lanetal., 2021)进行比对发现,167群东方蜜蜂样本全基因组重测序数据与参考基因组序列的平均匹配率为91.05%,平均测序深度为10.27×,数据质量达成后续分析要求。通过全基因组SNP calling,共获得137万个高质量的SNPs(表1)。

表1 基于全基因组重测序数据的青藏高原东缘、东南缘及邻近地区东方蜜蜂地理种群的SNPs数量与核苷酸多态性(θπ)Table 1 Numbers of SNPs and nucleotide polymorphisms (θπ) of the geographic populations of Apis ceranaon the eastern and southeastern edges of the Qinghai-Tibet Plateau and adjacent areasbased on whole-genome resequencing data
2.2 遗传分化
基于167群东方蜜蜂样本基因组SNPs,当假设样本中存在3个群体时(K=3),可区分出来自川西高原和藏南高原地理群体;当K=7时,来自滇北高原和川北高原地理群体依次从混合地理群体中区分出来(图1: A)。 这表明来自川西高原和川北高原的群体与藏南高原群体和滇北高原群体在遗传结构上具有明显的群体分化。主成分结果(图1: B)显示,167群东方蜜蜂样本被划分为川西高原、川北高原、藏南高原、滇北高原和其他地理群体。以上结果表明青藏高原东缘及东南缘分布着高原东方蜜蜂4个遗传分化群体,应该与该区域广阔且特殊的地理生境相关。

图1 基于SNPs的青藏高原东缘、东南缘及邻近地区东方蜜蜂遗传结构随分组组数(K)的变化(A)和主成分分析(B)Fig. 1 Genetic structure change with grouping number (K) (A) and principal component analysis (B) of Apis ceranaon the eastern and southeastern edges of the Qinghai-Tibet Plateau and adjacent areas based on SNPs种群信息见表1。For population information, see Table 1. WSichPl: 川西高原Western Sichuan Plateau; STibetPl: 藏南高原Southern Tibet Plateau; NYnanPl: 滇北高原Northern Yunnan Plateau; NSichPl: 川北高原Northern Sichuan Plateau. 下同The same below.
基于东方蜜蜂成对种群的遗传分化指数分析(表2)显示,11个种群的成对Fst值在0.0199~0.1443之间,平均0.0758。来自川西高原、川北高原、藏南高原和滇北高原的4个高原地理群体之间的Fst值(Fst=0.0917~0.1443,平均0.1178)高于非高原群体之间的Fst值(Fst=0.0219~0.0603,平均0.0411),这表明高原地理群体之间产生了较大的遗传分化,这与遗传结构分析(图1: A)和主成分分析(图1: B)结果一致。
不同种群间的最小遗传距离分析结果(表2)显示,相较于其他种群,DB和MEK与QLS之间的最小遗传距离值最小;BM和DQ与XSBN之间最小遗传距离最小;JZG与SNJ之间最小遗传距离最小。结合这些种群所属的地理位置,可以表明川西高原群体与川西山地群体具有更近的遗传关系;藏南高原、滇北高原群体与滇南群体具有更近的遗传关系;川北高原群体与秦巴群体具有更近的遗传关系。

表2 基于SNPs的青藏高原东缘、东南缘及邻近地区东方蜜蜂地理种群遗传分化指数(Fst)(上三角)和最小遗传距离(Nm)(下三角)Table 2 Pairwise genetic differentiation index (Fst)(above diagonal) and the minimum genetic distance (Nm) (below diagonal) of the geographic populations of Apis cerana on the eastern and southeastern edgesof the Qinghai-Tibet Plateau and adjacent areas based on SNPs
2.3 系统进化
基于SNPs构建的系统进化树(图2)显示,东方蜜蜂的青藏高原东缘及东南缘的高原群分为两个姐妹支,即东缘支和东南缘支,其中东南缘支包含藏南高原与滇北高原地理群体,东缘支则包含川西高原与川北高原地理群体。由此表明,对于东方蜜蜂高原群体而言,在地理位置上更接近的群体在亲缘关系上也更加接近。

图2 邻接法构建的基于SNPs的青藏高原东缘、东南缘的高原东方蜜蜂群的系统进化树(1 000次重复)Fig. 2 Phylogenetic tree of the plateau colonies of Apis cerana on the eastern and southeastern edges of the Qinghai-TibetPlateau constructed by neighbor-joining method based on SNPs (1 000 replicates)以西域黑蜂的SNPs为外群。The SNPs of Apis mellifera sinisxinyuan were used as the outgroup.
2.4 线粒体基因组单倍型
基于东方蜜蜂线粒体基因组单倍型分析,分别在川西高原组(MEK+DB+QLS)(图3: A)、川北高原组(JZG+SNJ) (图3: B)和藏南高原与滇北高原组(BM+DQ+XSBN) (图3: C)线粒体基因组中发现24, 16和21个单倍型。其中,川西高原组发现祖先单倍型Hap_15分布于QLS中,MEK与DB的单倍型为衍生单倍型;川北高原组中祖先单倍型Hap_6分布于SNJ中,JZG的单倍型为衍生单倍型;而在藏南高原与滇北高原组中XSBN的单倍型为主要单倍型,BM和DQ的单倍型为衍生单倍型。这为初步推断青藏高原东缘及东南缘高原东方蜜蜂来源提供了有效证据。

图3 青藏高原东缘、东南缘及邻近地区东方蜜蜂地理种群线粒体基因组单倍型网络图Fig. 3 Haplotype network of the mitochondrial genome of the geographic populations of Apis ceranaon the eastern and southeastern edges of the Qinghai-Tibet Plateau and adjacent areasA: 川西高原Western Sichuan Plateau; B: 川北高原Northern Sichuan Plateau; C: 藏南高原与滇北高原Southern Tibet Plateau and Northern Yunnan Plateau. Hap: 单倍型Haplotype.
2.5 环境适应性相关基因挖掘
在藏南高原组(BM+XSBN)基因组中发现71个潜在受选择区域和169个潜在受选择基因,主要富集在细胞脂质代谢过程、硫化合物生物合成等GO功能条目(图4),参与Wnt, FoxO, Notch和Hippo等KEGG通路(图5);在滇北高原组(DQ+XSBN)基因组中发现22个潜在受选择区域和53个潜在受选择基因,主要富集在防御反应调节、免疫反应调节等GO功能条目,参与FoxO等KEGG通路;在川西高原组(DB+QLS)基因组中发现40个潜在受选择区域和40个潜在受选择基因,主要富集在防御反应调节、DNA修复等GO功能条目,参与Toll, Imd, FoxO, MAPK和Hedgehog等KEGG通路;在川北高原组(JZG+SNJ)基因组中发现4个潜在受选择区域和10个潜在受选择基因,主要富集在神经肌肉的过程、大脑皮层发育等GO功能条目,参与线粒体自噬等KEGG通路。总体来说,这些受选择基因参与脂肪酸代谢、光转导、温度适应、卵巢发育等信号通路,但在功能富集区域又明显不同,这显示出不同高原东方蜜蜂在适应不同生境具有较大的基因选择差异。
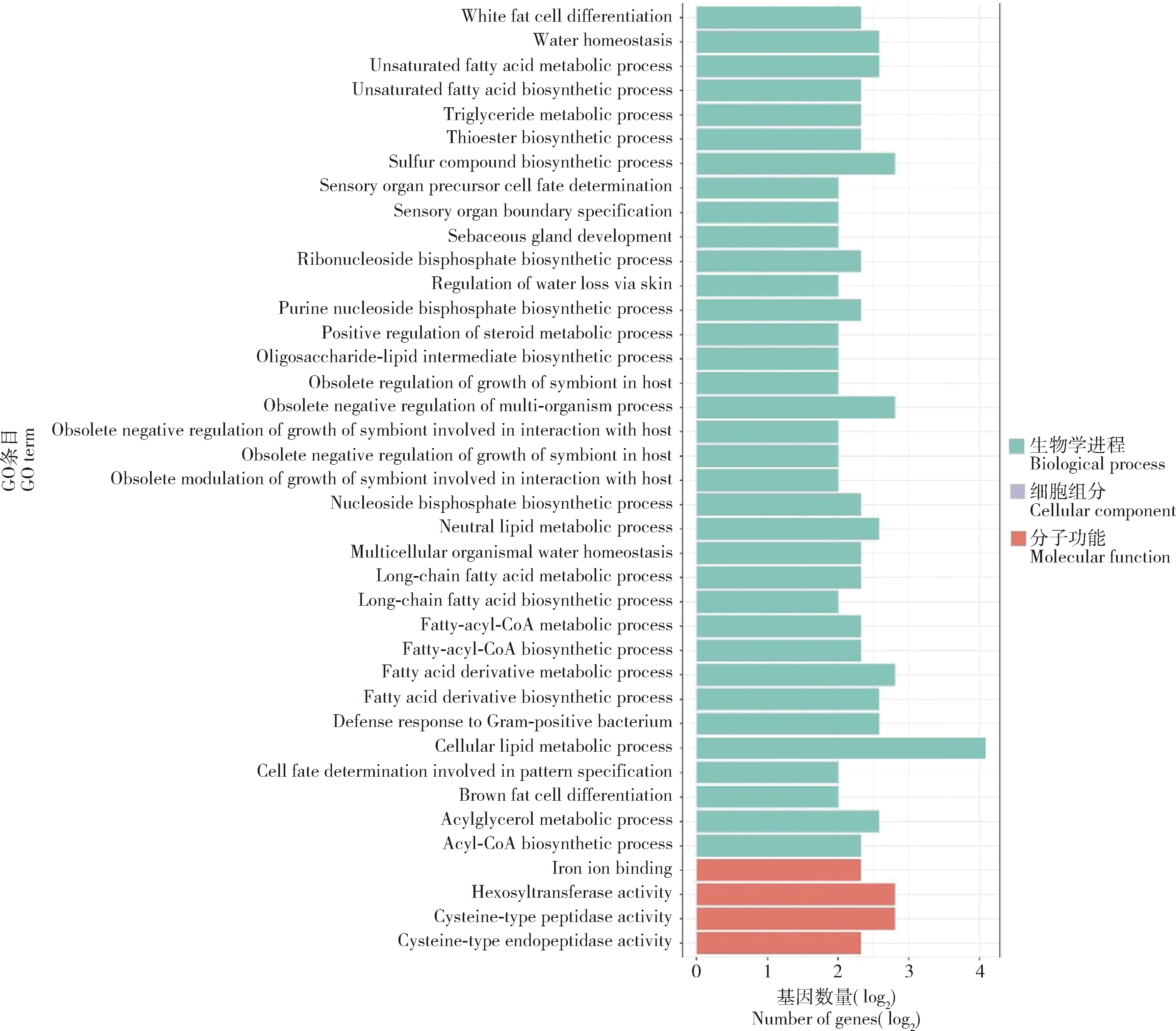
图4 藏南高原组(BM+XSBN)东方蜜蜂基因组中受选择基因的GO功能富集Fig. 4 GO functional enrichment of selected genes in the genomes of Apis ceranain the plateau group (BM+XSBN) of southern Tibet

图5 藏南高原组(BM+XSBN)东方蜜蜂基因组中受选择基因KEGG通路富集Fig. 5 KEGG pathway enrichment of selected genes in the genomes of Apis ceranain the plateau group (BM+XSBN) of southern Tibet
在滇南的藏南高原与滇北高原东方蜜蜂基因组中发现了5个共同的潜在受选择基因,分别为突触囊泡样糖蛋白2B基因(GenBank登录号: 107993575), UNC93样蛋白MFSD11基因(GenBank登录号: 107994554), 叉头盒蛋白O(FOXO)基因(GenBank登录号: 107993192), 胰岛素基因增强蛋白-1(ISL-1)基因(GenBank登录号: 107995439)以及一个未注释基因LOC107993577 (GenBank登录号: 107993577),其中ISL-1和FOXO,主要参与胰岛素分泌以应对细胞压力。
3 讨论
在本研究对青藏高原东缘及东南缘及邻近地区的11个地理种群的167群东方蜜蜂进行全基因组重测序数据分析,基于群体遗传结构和主成分分析发现,高原东方蜜蜂群分属川西高原、藏南高原、滇北高原、川北高原(图1: A, B),分布在青藏高原东缘及东南缘带上。遗传分化指数表明(表2),高原东方蜜蜂的遗传分化指数明显高于非高原地区的,且部分地区遗传分化指数也高于西方蜜蜂Apismellifera亚种间分化水平(平均Fst值约为0.1)(Wallbergetal., 2014),表明部分高原东方蜜蜂存在较大的遗传分化水平,达到了亚种分化水平。我们的研究结果也暗示着在青藏高原很可能存在更多的独特的地理遗传地理群体。
鉴于青藏高原地区多高山与峡谷,地处于这些地方的东方蜜蜂与邻近地区的东方蜜蜂的联系尚且未知。我们的研究基于各地理种群间遗传距离暗示了青藏高原东缘及东南缘东方蜜蜂与邻近地区东方蜜蜂的关系,研究表明藏南高原、滇北高原和滇南的东方蜜蜂具有更近的遗传关系,川西高原与川西山地的东方蜜蜂具有更近的遗传关系,而川北高原与秦巴神农架林区的东方蜜蜂具有更近的遗传关系(表2)。因此,结合本研究结果与此前相关研究(周丹银等, 2019; Jietal., 2020; Shietal., 2020),我们初步推测了青藏高原东缘及东南缘不同东方蜜蜂地理群体来源,即处于青藏高原东南缘的藏南高原、滇北高原东方蜜蜂来源于滇南,东缘的川西高原、川北高原东方蜜蜂分别来源于川西山地、秦巴,暗示了青藏高原东缘及东南缘东方蜜蜂是由邻近非高原东方蜜蜂的种群扩散后的地理隔离形成而产生的独立分化起源。
川西高原平均海拔约为3 740 m,主要属于温带-寒温带季风气候;滇北高原平均海拔约为3 380 m,气候类型与川西高原相似;藏南高原平均海拔超过4 500 m,主要属于亚热带山地湿润季风气候。气候多样性与地貌差异形成了青藏高原东缘及东南缘不同地理型的东方蜜蜂分化,而这种分化又与适应性进化具有一定的关联。我们基于对青藏高原东缘及东南缘高原东方蜜蜂与邻近非高原东方蜜蜂的选择性分析,发现潜在受选择基因参与多个信号通路(图5),其中,Wnt, MAPK, Notch以及Hippo信号通路在蜜蜂等昆虫的群体分化、胚胎形成、形态发生、成形盘发育和器官大小调节等发育过程中发挥重要作用(Deardenetal., 2006; Bolósetal., 2007; Komiya and Habas, 2008; Halder and Johnson, 2011; Wilsonetal., 2011; Yinetal., 2018)。Wnt, Notch和Hedgehog信号通路被认为与卵巢活动有关(Duncanetal., 2016; Chenetal., 2017; Chen Cetal., 2018),这可能显示出高原型较非高原型东方蜜蜂的卵巢活动更加强烈。Toll和Imd信号通路主要涉及包括蜜蜂在内的昆虫的免疫能力(Aneteetal., 2021)。Hippo信号通路在西方蜜蜂中被认为是适应寒冷气候的重要通路(Chen Cetal., 2016, 2018; Shietal., 2020),这显示出这些基因可能与高原东方蜜蜂耐寒性相关。除此之外,FoxO信号通路也被认为参与温度适应(Chen Cetal., 2016, 2018)。同时,我们还发现了潜在受选择基因参与代谢通路、光转导等相关通路,这些通路可能为高原东方蜜蜂适应不同生境提供了基础。基于来源于滇南的藏南高原与滇北高原东方蜜蜂的潜在受选择基因,我们发现了5个共同受选择基因。有研究表明ISL-1在调节细胞生命和胚胎发育中发挥重要作用(Pfaffetal., 1996),Evm.model.16.590 (ISL-1)和FOXO均参与调节胰岛素分泌以应对细胞压力(Jüngeretal., 2003; Guoetal., 2021),我们推测其为青藏高原东南缘的蜜蜂适应当地环境所需要。这对我们初步了解东方蜜蜂适应不同生境下的适应性基因情况提供了基础,为后续进行蜜蜂种质资源挖掘与遗传改良提供了参考。
猜你喜欢
今日农业(2022年3期)2022-11-16
军事文摘(2021年20期)2021-11-10
今日农业(2021年11期)2021-08-13
中国生殖健康(2020年4期)2020-12-09
中西医结合肝病杂志(2020年2期)2020-10-27
科学大众(2020年10期)2020-07-24
家教世界·创新阅读(2020年4期)2020-06-03
当代工人·精品C(2020年1期)2020-05-20
家教世界·创新阅读(2020年1期)2020-05-11
家教世界·创新阅读(2020年2期)2020-04-07
