
固肾定喘丸多指标成分一测多评法的建立及质量评价Δ
2022-09-30 14:26李志平高雨秋王加良侯甲福牡丹江医学院附属红旗医院药学部黑龙江牡丹江570牡丹江医学院药学院黑龙江牡丹江570
中国药房 2022年18期
张 颖,李志平,高雨秋,王加良,侯甲福(.牡丹江医学院附属红旗医院药学部,黑龙江 牡丹江 570;.牡丹江医学院药学院,黑龙江 牡丹江 570)
固肾定喘丸由肉桂、熟地黄、车前子、盐补骨脂、附片、盐益智仁等13味中药饮片加工而成,方中盐补骨脂温肾助阳、纳气平喘,为君药;附片和肉桂补肾阳、固肾定喘,盐益智仁和金樱子肉温,补脾肾,合为臣药;熟地黄、山药、茯苓、牡丹皮和泽泻滋补肾阴、渗湿热,车前子和牛膝利水渗湿、补肝肾,合为佐药;砂仁化湿开胃、温脾止泻,为使药。固肾定喘丸可温肾纳气、健脾化痰,临床主要用于治疗肺脾气虚、肾不纳气所致的咳嗽和气喘,亦可用于慢性支气管炎、肺气肿、支气管哮喘而引起的咳嗽和气喘。固肾定喘丸收载于2020年版《中国药典》(一部)[1],其质量标准及文献报道[2]仅对君药盐补骨脂中补骨脂素进行了定量分析。对于所含化学成分复杂、临床作用机制不清晰的中药复方制剂来说,检测指标过于单一,对体现中药制剂的整体质量、保证其产品质量一致性存在一定的局限。
近年来,一测多评(quantitative analysis of multicomponents by single marker,QAMS)法的定量分析越来越多地应用于中成药复方制剂质量评价中[3]。化学模式识别分析可通过统计学或数学方法,挖掘复杂数据间存在的内在函数关系,现已广泛应用于中药多维信息的综合分析中[4]。本实验参考中药质量标志物确认依据,选取纯度较高、质量稳定、价廉易得的补骨脂素为内参物,采用QAMS法对固肾定喘丸中君药盐补骨脂特征成分补骨脂素、异补骨脂素、新补骨脂异黄酮和补骨脂二氢黄酮,臣药肉桂药效成分肉桂酸和桂皮醛,佐药熟地黄和车前子主要成分大车前苷、毛蕊花糖苷、异毛蕊花糖苷和木通苯乙醇苷B共10个成分的含量进行检测,建立固肾定喘丸多指标成分一测多评法,同时利用化学模式识别分析方法对不同批次样品的检测结果进行评价,以期完善并提高固肾定喘丸的质量控制手段,确保产品质量稳定和临床疗效的一致性。
1 材料
1.1 主要仪器
本研究所用主要仪器有1200型高效液相色谱(HPLC)仪(美国Agilent公司)、2695型HPLC仪(美国Waters公司)、MS105DU型电子天平(瑞士Mettler Toledo公司)、SB-5200DTDN型超声波清洗机(宁波新芝生物科技有限公司)等。
1.2 主要药品与试剂
固肾定喘丸(水蜜丸,国药准字Z44020906)购自广州白云山敬修堂药业股份有限公司,批号分别为L02008、L03011、L03016、L04018、L06035、L06041、L08050、M04001、M12021、M12050、Y04007、Y10010、T12001、F08027和H10028,编号依次为S1~S15。肉桂酸对照品(批号110786-201604,纯度98.8%)、桂皮醛对照品(批号110710-202022,纯度99.5%)、大车前苷对照品(批号111914-202105,纯度96.0%)、毛蕊花糖苷对照品(批号111530-201914,纯度95.2%)、木通苯乙醇苷B对照品(批号111910-201604,纯度98.2%)、补骨脂素对照品(批号110739-201918,纯度99.6%)、异补骨脂素对照品(批号110738-202016,纯度99.4%)、新补骨脂异黄酮对照品(批号520052-201401,纯度99.6%)和补骨脂二氢黄酮对照品(批号520053-201401,纯度99.4%)均购自中国食品药品检定研究院;异毛蕊花糖苷对照品(批号PRF9010243,纯度97.0%)购自成都普瑞法科技开发有限公司;乙腈、甲醇均为色谱纯,其余试剂均为分析纯。
2 方法与结果
2.1 色谱条件
以Agilent SB-C18(250 mm×4.6 mm,5 μm)为色谱柱,以乙腈(A)-0.2%冰醋酸溶液(B)为流动相进行梯度洗脱(0~11 min,25%A;11~20 min,25%A→40%A;20~35 min,40%A→50%A;35~59 min,50%A→75%A;59~70 min,75%A→25%A);柱温为30℃;流速为1.0 mL/min;检测波长分别为290 nm(0~20 min,检测肉桂酸和桂皮醛)[5-6]、330 nm(20~35 min,检测大车前苷、毛蕊花糖苷、异毛蕊花糖苷和木通苯乙醇苷B)[7-9]和246 nm(35~70 min,检测补骨脂素、异补骨脂素、新补骨脂异黄酮和补骨脂二氢黄酮)[10-11];进样量为10µL。
2.2 溶液的制备
2.2.1 混合对照品溶液 精密称取肉桂酸、桂皮醛、大车前苷、毛蕊花糖苷、异毛蕊花糖苷、木通苯乙醇苷B、补骨脂素、异补骨脂素、新补骨脂异黄酮和补骨脂二氢黄酮对照品适量,用甲醇溶解制成质量浓度分别为0.198、0.472、0.296、2.130、0.414、0.136、1.408、0.960、0.572、0.538 mg/mL的混合对照品贮备液。将混合对照品贮备液用甲醇稀释20倍制得混合对照品溶液(上述成分质量浓度分别为9.9、23.6、14.8、106.5、20.7、6.8、70.4、48.0、28.6、26.9 µg/mL)。
2.2.2 供试品溶液 取固肾定喘丸适量,研细,取约3 g,精密称定,置具塞锥形瓶中,精密加入甲醇25 mL,称定质量后,水浴回流提取60 min,放冷,用甲醇补足减失质量,摇匀,经0.45 μm微孔滤膜过滤,取续滤液,即得。
2.2.3 阴性供试品溶液 取按固肾定喘丸标准处方和制法制备的缺肉桂、缺盐补骨脂、缺熟地黄和车前子的3种阴性供试品各适量,按“2.2.2”项下方法制得相应阴性供试品溶液。
2.3 方法学考察
2.3.1 专属性考察 取混合对照品溶液、供试品溶液和阴性供试品溶液,按“2.1”项下色谱条件进样测定,记录色谱图(图1)。结果显示,供试品溶液中各待测成分与相邻色谱峰分离良好(分离度均不小于1.5),理论板数按各成分色谱峰计均不小于5 500,阴性供试品对测定无干扰。
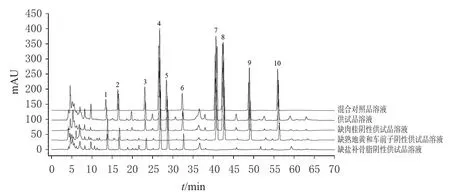
图1 固肾定喘丸定量分析的HPLC图
2.3.2 线性关系考察 精密吸取混合对照品贮备液0.1、0.2、0.5、1.0、2.0、5.0 mL,置于不同的20 mL量瓶中,用甲醇稀释制得6个系列质量浓度的混合对照品溶液,按“2.1”项下色谱条件进样测定,记录峰面积。以各待测成分质量浓度为横坐标(X)、峰面积为纵坐标(Y)进行线性回归,结果见表1。

表1 固肾定喘丸中10个待测成分的回归方程和线性范围
2.3.3 精密度试验 取供试品溶液(编号S1),按“2.1”项下色谱条件连续进样6次,记录峰面积。结果显示,肉桂酸等10个待测成分峰面积的RSD均小于2.00%(n=6),表明方法精密度良好。
2.3.4 重复性试验 精密称取同一批固肾定喘丸样品(编号S1)6份,分别按“2.2.2”项下方法制备供试品溶液,按“2.1”项下色谱条件进样测定,记录峰面积并用外标法计算含量。结果显示,肉桂酸等10个待测成分含量的RSD均小于2.00%(n=6),表明方法重复性良好。
2.3.5 稳定性试验 取供试品溶液(编号S1),分别在室温下放置0、2、4、6、12、24 h时按“2.1”项下色谱条件进样测定,记录峰面积。结果显示,肉桂酸等10个待测成分峰面积的RSD均小于2.00%(n=6),表明供试品溶液在室温下放置24 h内稳定性良好。
2.3.6 加样回收率试验 取已知待测成分含量的固肾定喘丸样品(编号S1)适量,研细,分别取9份,每份精密称定1.5 g,均分成3组,每组分别精密加入混合对照品溶液(肉桂酸、桂皮醛、大车前苷、毛蕊花糖苷、异毛蕊花糖苷、木通苯乙醇苷B、补骨脂素、异补骨脂素、新补骨脂异黄酮和补骨脂二氢黄酮的质量浓度分别为0.107、0.351、0.232、1.427、0.278、0.072、0.931、0.709、0.435 和0.379 mg/mL)0.8、1.0、1.2 mL,按“2.2.2”项下方法制备供试品溶液,再按“2.1”项下色谱条件进样测定,记录峰面积并计算加样回收率。结果显示,肉桂酸等上述10个待测成分的平均加样回收率为96.98%~100.09%,RSD均小于2.00%(n=9),表明方法准确度良好。
2.4 QAMS法的建立
2.4.1 相对校正因子的计算 用对照品质量浓度与峰面积之比计算各待测成分的相对校正因子(relative correction factor,RCF),即RCF=(Wk×As)/(Ws×Ak)(式中W和A分别为质量浓度和峰面积,下标k和s为其他待测成分和内参物)。按“2.1”项下色谱条件进样测定“2.3.2”项下6个系列质量浓度的混合对照品溶液,以补骨脂素为内参物,计算得肉桂酸、桂皮醛、大车前苷、毛蕊花糖苷、异毛蕊花糖苷、木通苯乙醇苷B、异补骨脂素、新补骨脂异黄酮和补骨脂二氢黄酮的RCF分别为2.818 7、1.567 8、2.202 3、1.227 0、1.744 4、4.466 7、0.970 0、1.084 8和1.257 7,RSD均小于2.00%(n=6)。
2.4.2 RCF耐用性试验 精密吸取“2.2.1”项下混合对照品溶液,按“2.1”项下色谱条件进样测定,记录峰面积,分别考察不同HPLC仪(Waters 2695型、Agilent 1200型,下同)、不同色谱柱[Agilent SB-C18、Phenomenex SuperLu C18、Welch Ultimate XB C18,规格均为(250 mm×4.6 mm,5 μm),下同]、不同流速(0.8、1.0、1.2 mL/min)及不同柱温(25、30、35℃)对所建立的RCF的影响。结果显示,不同仪器及不同色谱柱下肉桂酸等上述9个待测成分的平均RCF分别为2.816 4、1.571 5、2.204 5、1.222 4、1.743 5、4.463 6、0.968 1、1.079 0和1.254 0,不同流速下的平均RCF分别为2.8147、1.5606、2.2083、1.2278、1.7462、4.468 9、0.971 1、1.081 7和1.259 2,不同柱温下的平均RCF 分别为 2.813 1、1.564 3、2.218 0、1.225 6、1.742 6、4.466 0、0.963 8、1.082 9和1.255 0,RSD均小于2.00%(n=9),表明RCF耐用性良好。
2.4.3 色谱峰定位 采用相对保留时间值(relative retention time,RRT)法对待测成分色谱峰进行定位,精密吸取“2.2.1”项下混合对照品溶液,按“2.1”项下色谱条件进样测定,以补骨脂素为内参物,考察不同仪器和不同色谱柱对肉桂酸等上述9个待测成分的RRT的影响。结果显示,9个待测成分的RRT分别为0.332 0、0.405 2、0.563 9、0.653 8、0.699 9、0.794 8、1.041 9、1.203 2 和1.380 6,RSD均小于2.00%(n=6)。
2.5 含量测定
取15批固肾定喘丸样品(编号S1~S15),分别按“2.2.2”项下方法制备供试品溶液,按“2.1”项下色谱条件进样测定,记录峰面积,分别采用外标法和QAMS法计算样品中上述10个待测成分的含量(表2)。采用SPSS 26.0软件进行t检验,比较2种方法检测结果的差异。结果显示,2种方法的检测结果差异无统计学意义(P>0.05)。

表2 15批固肾定喘丸中10个待测成分含量测定结果(mg/g,n=3)
2.6 固肾定喘丸化学模式识别分析方法的建立
2.6.1 聚类分析 将“2.5”项下15批固肾定喘丸中10个待测成分QAMS法含量数据导入SPSS 26.0软件进行聚类分析(cluster analysis,CA)。结果显示,当欧氏距离为15时,15批样品聚为3类,S1~S7为第Ⅰ类,S8~S10为第Ⅱ类,S11~S15为第Ⅲ类。
2.6.2 主成分分析 将“2.5”项下15批固肾定喘丸中10个待测成分QAMS法含量数据导入SPSS 26.0软件进行主成分分析(principal component analysis,PCA)。结果显示,前2个主成分特征值分别为6.391和2.654,对主成分的方差贡献率分别为63.905%和26.541%,累计方差贡献率为90.446%(大于85%),表明选取前2个主成分即可代表固肾定喘丸90.446%的信息量。第一主成分的信息来自肉桂酸、大车前苷、毛蕊花糖苷、异毛蕊花糖苷、木通苯乙醇苷B、新补骨脂异黄酮和补骨脂二氢黄酮的综合,第二主成分的信息来自桂皮醛、补骨脂素和异补骨脂素的综合。同时应用SIMCA 14.1软件建立PCA模型,得15批固肾定喘丸样品的PCA得分图(图2)。从图2可以看出,15批样品聚为3类,与CA结果一致。
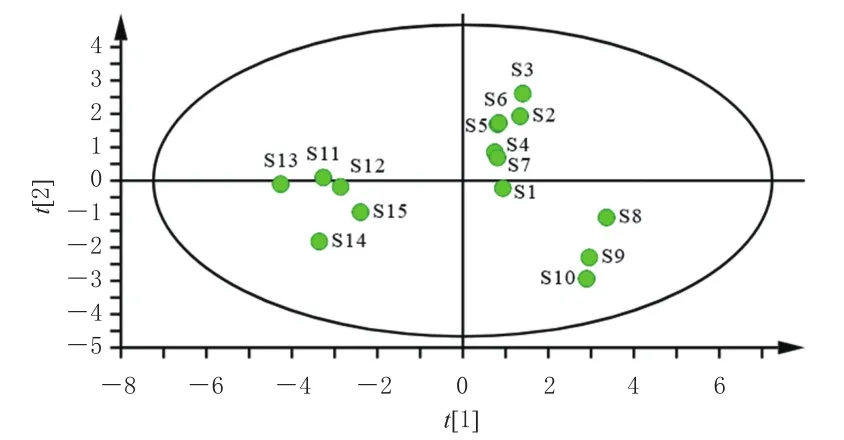
图2 15批固肾定喘丸样品的PCA得分图
2.6.3 偏最小二乘法-判别分析 将“2.5”项下15批固肾定喘丸中10个待测成分QAMS法含量数据导入SIMCA 14.1软件,运行偏最小二乘法-判别分析(partial least squares discrimination analysis,PLS-DA)程序,得图3。由图3可知,模型的累积解释能力参数(R2X、R2Y)分别为0.917和0.851,预测能力参数(Q2)为0.789,均大于0.5,说明所建立的模型稳定可靠、预测能力强[12],可用于区分不同批次的固肾定喘丸。
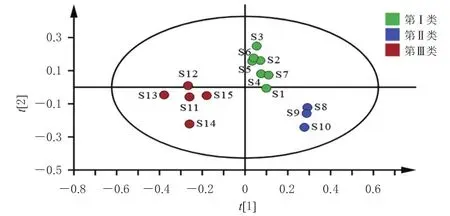
图3 15批固肾定喘丸样品的PLS-DA模型得分图
对建立的PLS-DA模型进行200次置换检验(图4),结果显示,R2拟合直线Y轴截距为0.076 7,小于0.3,表明所建立的模型结果可靠;Q2拟合直线Y轴截距为-0.296,小于0.05,表明所建立的模型不存在过度拟合,可有效判别分析15批固肾定喘丸的质量差异。根据变量重要性投影(variable importance in projection,VIP)值筛选影响固肾定喘丸质量的标志性成分,结果显示,补骨脂素、毛蕊花糖苷、桂皮醛和异补骨脂素是影响该药质量的标志性成分(VIP值均大于1.0)。
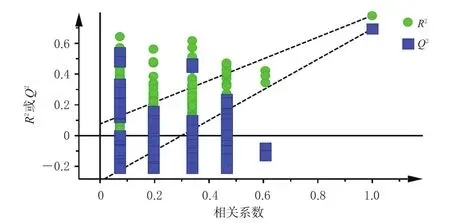
图4 15批固肾定喘丸样品的PLS-DA模型置换检验图
3 讨论
本实验在制备供试品溶液时,考察了不同提取方法(超声提取和水浴回流提取)、不同提取溶剂(水、甲醇和乙醇)、不同提取时间(30、60、90 min)对固肾定喘丸样品中10个待测成分提取率的影响及杂质干扰情况。结果显示,水浴回流提取效率较高;溶剂为甲醇时,10个待测成分的响应值较大;提取时间为60 min时,提取效率最高,同时杂质最少。综合以上条件,选取甲醇水浴回流提取60 min为最佳提取方式。
本实验在筛选流动相时,首先以甲醇-水、乙腈-水为流动相,发现以甲醇-水为流动相时,检测用时较长,且数个色谱峰分离度达不到要求;以乙腈-水为流动相时,毛蕊花糖苷、补骨脂素和补骨脂二氢黄酮色谱峰出现拖尾现象,考虑用酸类溶液加以改善。通过对比乙腈-0.1%磷酸溶液[13]、乙腈-0.1%甲酸溶液[14]、乙腈-0.2%冰醋酸溶液[15]为流动相时10个待测成分的分离效果,结果显示,以乙腈-0.2%冰醋酸溶液为流动相时,10个待测成分色谱峰均可达到基线分离且峰形较好。因此,选择乙腈-0.2%冰醋酸溶液为流动相对固肾定喘丸中10个待测成分同时进行含量测定。
外标法和QAMS法所得10个待测成分含量测定结果差异无统计学意义(P>0.05),表明本研究建立的QAMS法较为合理。从CA、PCA和PLS-DA结果可以看出,15批固肾定喘丸样品可聚为3类,君药所含成分补骨脂素和异补骨脂素,臣药所含成分桂皮醛及佐药所含成分毛蕊花糖苷是影响固肾定喘丸产品质量的潜在标志性成分。综上所述,本研究所建立的QAMS法多指标成分定量控制及化学模式识别分析可用于固肾定喘丸的质量评价。
猜你喜欢
航天电子对抗(2022年4期)2022-10-24
草业科学(2022年9期)2022-10-21
中草药(2022年11期)2022-05-31
中草药(2022年9期)2022-05-06
河南农业·综合版(2022年2期)2022-03-18
河南农业(2022年2期)2022-03-14
核农学报(2022年1期)2022-03-11
河南农业·综合版(2021年7期)2021-08-23
农业与技术(2017年17期)2017-09-30
中国医药导报(2011年27期)2011-12-31
