
X连锁低磷血症性佝偻病剪接位点突变及表型分析
2022-11-16 03:35杨一凡王亭刘世国李冰
青岛大学学报(医学版) 2022年5期
杨一凡 王亭 刘世国 李冰

[摘要] 目的 分析1例X连锁低磷血症性佝偻病(XLH)病人遗传学改变及其基因型表型。
方法 对1例散发性低磷血症性佝偻病病人进行全外显子组测序(WES)分析,找出致病基因突变位点,结合病人临床表现进行基因型和表型的分析。对先证者及其同胞进行Sanger测序验证。
结果 实验室检查显示,先证者血清磷降低,碱性磷酸酶升高,甲状旁腺激素升高;X射线及磁共振成像检查显示,病人下肢严重内翻畸形,胸椎管狭窄,腰椎间盘突出及胸腰椎退行性病变。测序结果显示,先证者PHEX基因的第15内含子发现一个异常剪接位点的杂合突变c.1646-2A>G,其同胞未发现该位点的突变。因此,先证者被诊断为XLH。
结论 本研究发现了1例XLH病人PHEX基因剪接位点突变,有助于了解和完善中国XLH病人的遗传学基础。
[关键词] 家族性低血磷性佝偻病;全外显子组测序;X染色体上内肽酶同源磷调节基因;DNA突变分析
[中图分类号] R596.1
[文献标志码] A
[文章编号] 2096-5532(2022)05-0639-04
doi:10.11712/jms.2096-5532.2022.58.126
[开放科学(资源服务)标识码(OSID)]
[网络出版] https://kns.cnki.net/kcms/detail/37.1517.r.20220713.0951.002.html;2022-07-13 17:12:23
SPLICE SITE MUTATION AND PHENOTYPE IN X-LINKED HYPOPHOSPHATEMIC RICKETS: A CASE ANALYSIS
YANG Yifan, WANG Ting, LIU Shiguo, LI Bing
(Department of Biology, School of Basic Medicine, Qingdao University, Qingdao 266071, China);
[ABSTRACT] Objective To analyze the genetic change, genotype, and phenotype of a patient with X-linked hypophosphatemic rickets (XLH).
Methods A case of sporadic hypophosphatemic rickets was analyzed by whole exome sequencing to identify the mutation site of the disease gene, and the genotyping and phenotypic analysis were performed in combination with the clinical manifestations of the patient. The proband and her siblings were validated by Sanger sequencing.
Results Laboratory examinations for the proband showed a decrease in phosphorus and an increase in alkaline phosphatase and parathyroid hormone in the serum. Radiological examinations (X-ray, and magnetic resonance imaging) revealed severe varus deformity of the lower limbs, thoracic spinal stenosis, lumbar disc herniation, and thoracolumbar degenerative disease. Sequencing results showed that an abnormal heterozygous mutation at the splice site c.1646-2A>G was identified at the intron 15 of the PHEX gene in the proband, which was not found in her siblings. Therefore, the proband was diagnosed as having XLH.
Conclusion In this study, a splice site mutation in the PHEX gene has been identified in a patient with XLH, which is helpful to understand and improve the genetic basis of XLH patients in China.
[KEY WORDS] familial hypophosphatemic rickets; whole exome sequencing; phosphate-regulating gene with homologies to endopeptidase on the X chromosome; DNA mutational analysis
低磷血癥性佝偻病是一种具有高度临床和遗传异质性的骨代谢障碍性疾病,其临床表现为生长缓慢、负重肢体弯曲,一般在1~2岁后表现明显,常伴有肾脏磷酸盐重吸收缺陷和异常骨矿化[1-3]。X-连锁低磷血症性佝偻病(XLH,OMIM#307800)是其中最常见的类型,发病率约为 1/20 000[4]。XLH是由于X染色体上内肽酶同源磷调节基因(PHEX,OMIM#300550,NM_000444.6)失活性突变引起的[5]。PHEX基因位于Xp22.1上,由22个外显子组成,全长243 kb[6-7]。常规技术检查(包括放射技术检查和电生理检查)对XLH诊断有辅助性作用,但由于XLH具有临床和遗传异质性,无法根据临床数据确定基因型与表型的相关性。本研究通过全外显子组测序(WES)和Sanger测序验证,结合病人的实验室和影像学检查结果,从1个XLH中国家系中鉴定出1个PHEX基因的异常剪接突变。现将结果报告如下。
1 资料和方法
1.1 研究对象
病人来自山东省的一个患有XLH的中国家庭。先证者为女性,47岁(图1中Ⅱ3),根据临床表现、影像学检查和实验室检查,在青岛大学附属医院临床诊断为XLH。其他家系成员均无此病(图1)。本研究经青岛大学附属医院伦理委员会批准,并获得家系成员的知情同意。
正方形表示男性,圆圈表示女性;黑色符号表示受影响的成员;黑色箭头表示家族中的先证者。
1.2 实验室和影像学检查
采集先证者空腹静脉血,应用日立7600全自动生化分析仪测定血钙、磷和碱性磷酸酶(ALP)浓度。用ECLIA Elecsys全自动分析仪(E170,德国曼海姆罗氏诊断公司)测定血清1,25-双羟维生素D(1,25(OH)2D)和甲状旁腺激素(PTH)水平。用3.0 Tesla临床扫描仪(GE HDX 3.0 T MR系统,GE Medical System,Milwaukee,USA)对病人进行腰椎和胸椎磁共振成像(MRI)检查,并与之前在同一家医院、使用相同的MRI技术和相同的临床扫描仪检查、年龄和性别匹配的健康个体50名进行比较。同时进行X线检查。
1.3 突变基因检测
1.3.1 基因组DNA提取 分别采集该家系Ⅱ代成员的血液样本2 mL,使用Qiagen DNA试剂盒提取DNA,严格按照试剂盒说明书操作。
1.3.2 WES分析 对先证者(Ⅱ3)进行WES分析。打断基因组DNA,完成DNA片段化和接头连接,制备cDNA文库。应用Aglient 2100检验合格的cDNA文库,将经过富集的DNA应用Illumina HISeq 2000测序仪进行双末端测序,平均测序深度达96.2×,目标区域97.67×,覆盖率为98%。去除接头污染和低质量数据后,对测序结果进行生物信息学分析,筛选结果应用MaxEntScan、Spliceman对可能有害的突变进行预测,从而发现该家系致病的突变基因。
1.3.3 Sanger测序 将WES所测得的高致病突变,在该家系第Ⅱ代样本中进行Sanger测序,以验证WES结果的准确性。使用Primer express 3.0软件设计引物(引物序列F:CATGTATGAAGGAGCCCAGCTT;R:CCTGGTAACAAGGATCAGAAAACA),对检出突变所在外显子序列进行PCR扩增。PCR反应体系为50.0 μL,包括:Master Mix 21.5 μL,上下游引物各1.0 μL,模板DNA 1.5 μL。PCR反应条件为:98 ℃预变性2 min;98 ℃变性10 s,56 ℃退火10 s,72 ℃延伸5 s,共35个循环;72 ℃终延伸2 min。扩增完成后,PCR产物经浓度为10 g/L的琼脂糖凝胶电泳鉴定,对扩增条带明亮且單一者使用ABI 3730XL测序仪(Applied Biosystems 3730-XL)进行测序。用BioEdit(Borland,V7.0.1)程序对DNA序列进行分析,并与GenBank数据库中的参考序列进行比较。然后应用聚合物鉴定突变位点,比对人类基因突变数据库(HGMD,http://www.hgmd.cf.ac.uk/ac/index.php)鉴定新突变。
2 结 果
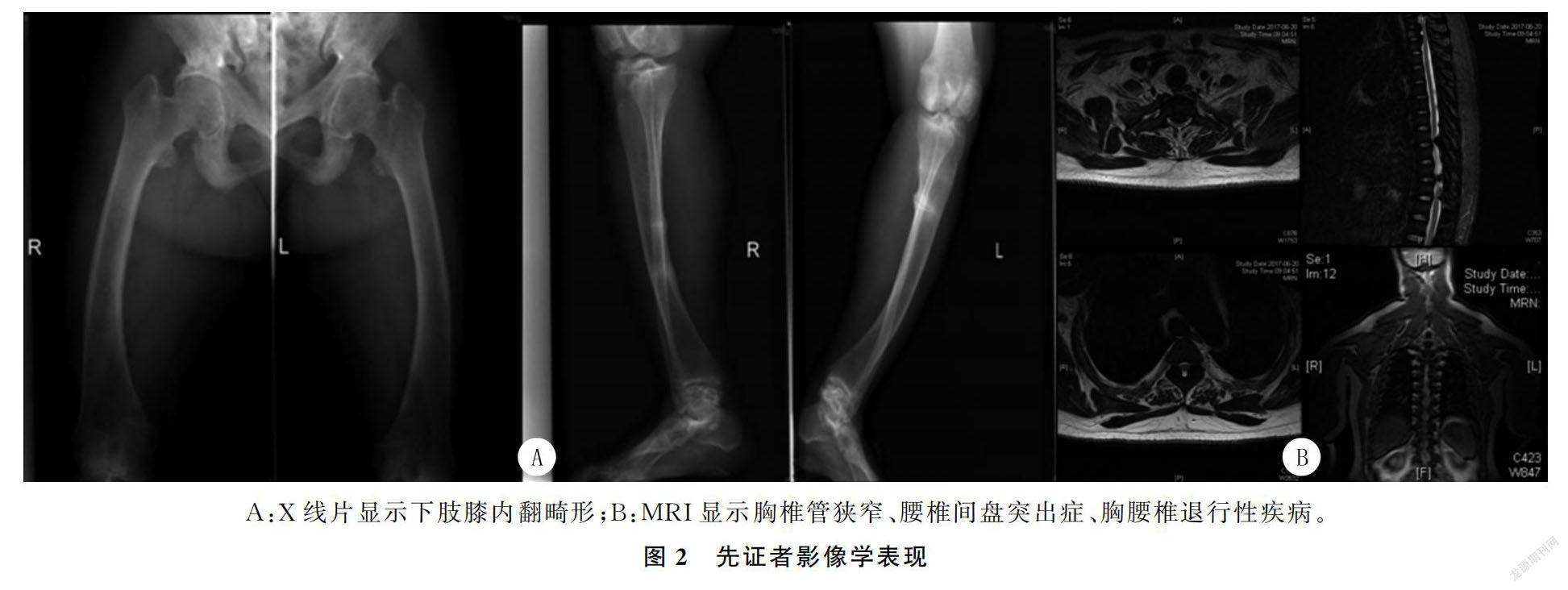
2.1 XLH家系临床特点和实验室检查先证者,女,47岁。父母非近亲结婚,G3P3,正常自然阴道分娩。出生体质量2.4 kg,初次啼哭无延迟。临床表现为身材矮小(身高140 cm,体质量65 kg,体质量指数(BMI)33.16 kg/m2),下肢膝内翻严重畸形,牙齿发育不良。临床主要症状为步态异常,腰痛10年,下肢麻木2年。其他家庭成员无相关临床症状。先证者实验室检查结果显示:血钙2.16 mmol/L(正常范围2.08~2.60 mmol/L),血磷0.5 mmol/L(正常范围0.8~1.6 mmol/L),ALP 133 U/L(正常范围15~112 U/L),PTH 67 ng/L(正常范围15~65 ng/L),1,25(OH)2D 21.24 μg/L(正常范围20~35 μg/L)。其他家系成员的临床表现和实验室结果均正常。先证者X线检查显示严重的下肢内翻畸形(图2A);MRI检查:胸椎管狭窄、腰椎间盘突出、胸腰椎退行性病变(图2B),全身松质骨数量增加,骨密度增高,骨盆韧带骨化。其他50名健康个体影像学检查均无异常。
2.2 突变基因的WES分析及Sanger测序验证
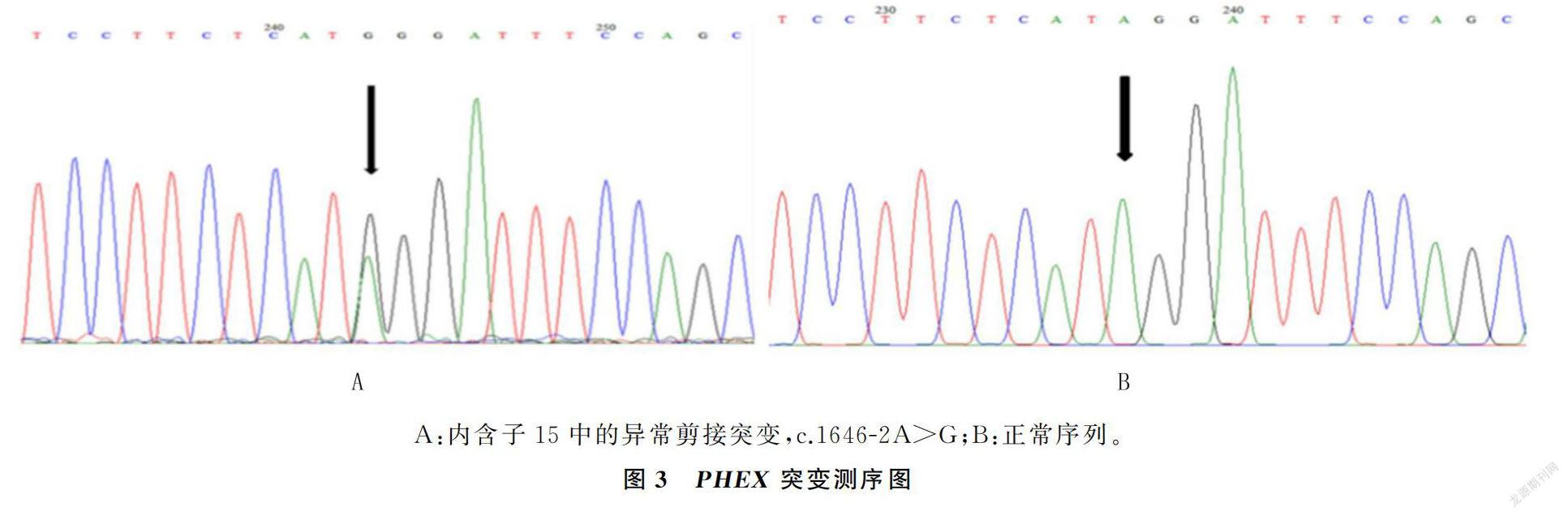
WES分析显示,先证者存在1个PHEX基因剪接位点杂合突变(c.1646-2A>G,NM_000444.
6),即该基因编码cDNA的第1646位点(第15内含子)上游的第2个碱基由腺嘌呤(A)变为鸟嘌呤(G),导致剪接位点突变,从而引起编码蛋白功能异常。MaxEntScan(http://hollywood.mit.edu/bur-gelab/maxent/Xmaxentscan_scoreseq.html)分析显示,Maximum Entropy Model(MAXENT)-6.23,First-order Markov Model(MM)-5.56,Weight Matrix Model(WMM)-3.09,对应的野生型评分分别为11.09、11.88、13.81;Spliceman指数显示62%。该突变可能会影响mRNA剪接,从而影响编码蛋白功能,最终导致疾病表现。
对WES分析显示的高度可疑致病位点c.1646-2A>G进行Sanger测序验证,结果表明,先证者携带该致病突变,而家系中其他两位正常成员(Ⅱ1、Ⅱ2)未发现该基因的异常。此外,在中国山东的100名无关健康对照者中也未发现突变位点。因此,先证者可被诊断为XLH。
3 讨 论
XLH是低磷血症性佝偻病中最常见的类型,因为病人维生素D吸收相对无效,因此XLH病人的临床表现与大多数佝偻病病人的临床表现不同[9]。XLH病人实验室检查表现为低磷血症、ALP升高、血钙正常、1,25(OH)2D正常或偏低、PTH正常或偏高[4]。XLH病人在儿童发育早期由于肾脏磷酸盐渗漏导致血液中磷酸盐水平降低和骨矿化缺陷而对维生素D治疗产生抵抗力;随着年龄增长,病人会出现自发性牙齿脓肿、广泛骨痛和关节病等[5-7]。虽然患有XLH的病人临床表现不尽相同,但其特点均为佝偻骨畸形、身材矮小和牙齿畸形等[3]。
PHEX的失活性突变可致磷调节因子纤维生长因子23(FGF23)升高,减少肾小管磷的重吸收,抑制活性维生素D的生成,导致骨骼矿化障碍[9]。因此,PHEX基因的功能丧失伴随骨骼矿化缺陷、FGF23的合成和分泌增强。迄今为止,在XLH病人中已发现约460个PHEX突变,国内也有相关病例报道[10-12]。然而,PHEX失活导致XLH骨骼和肾脏异常的确切机制尚未完全阐明。
PHEX基因如发生病理性变异可导致低磷血症性佝偻病,通常以X连锁显性的方式遗传:男性病人可将病理性变异遗传给女儿,女儿患病率为100%,因为男性通常只有一个X染色体的半合子,所以X连锁显性疾病在男性中的表达率通常高于女性[13-15]。女性病人有50%的可能将病理性变异遗传给子女,但通常女性病人的症状较轻,无明显的骨骼发育畸形,只表现为低磷血症[16]。
对散发性病人,PHEX基因突变的分子诊断对于确认XLH的临床诊断,进行遗传咨询和促进产前干预具有重要意义。国内外研究显示,XLH病人在出现生长障碍及佝偻病体征前的婴儿期开始治疗,其身高改善及病情减轻较1岁后开始治疗者效果好[17]。XLH病人的子女应在出生后3个月内仔细追踪,尽早治疗。
2014年YUE等[18]报道了9个中国汉族无关家系(其中有3个散发性病例)的16例病人(5例男性,11例女性),12例病人有膝内翻的表现,但严重程度不一;4例病人存在牙齿发育异常,其中2例病人有不同程度的牙齿脱落。对相关研究报道的病例进行比较显示,不同种族的相同基因突变会导致不同的临床特征[1,18]。
本研究通过对1例中国XLH病人的WES分析及Sanger测序验证,结合病人的临床表现、实验室和影像学检查结果,确定了一个影响RNA转录本的致病基因突变,该突变位于PHEX基因内含子15剪接受体位点(c.1646-2A>G)。随着对XLH遗传学分子基礎认识的加深和我国XLH病人数量的增加,将有望阐明PHEX基因复杂的基因型-表型关系和功能,有助于XLH的早期分子诊断和治疗。本研究为深入了解XLH的病理生理机制,更准确迅速地诊断该遗传性出生缺陷疾病奠定基础。今后需要对更多的中国XLH病人进行研究,以阐明PHEX突变的分布和频率以及中国XLH病人的基因型-表型关系。
[参考文献]
[1]ROWE P S. The molecular background to hypophosphataemic rickets[J]. Archives of Disease in Childhood, 2000,83(3):192-194.
[2]HOLM I A, NELSON A E, ROBINSON B G, et al. Mutational analysis and genotype-phenotype correlation of the PHEX gene in X-linked hypophosphatemic rickets[J]. The Journal of Clinical Endocrinology and Metabolism, 2001,86(8):3889-3899.
[3]PAVONE V, TESTA G, GIOITTA IACHINO S, et al. Hypophosphatemic rickets: etiology, clinical features and treatment[J]. European Journal of Orthopaedic Surgery & Traumatology: Orthopedie Traumatologie, 2015,25(2):221-226.
[4]QUINLAN C, GUEGAN K, OFFIAH A, et al. Growth in PHEX-associated X-linked hypophosphatemic rickets: the importance of early treatment[J]. Pediatric Nephrology (Berlin, Germany), 2012,27(4):581-588.
[5]李丽,徐杨,戚露月,等. X连锁显性低血磷性佝偻病患者的临床特征与致病基因鉴定[J]. 中华骨质疏松和骨矿盐疾病杂志, 2019,12(4):336-346.
[6]TIEDER M, MODAI D, SHAKED U, et al. “Idiopathic” hypercalciuria and hereditary hypophosphatemic rickets. Two phenotypical expressions of a common genetic defect[J]. The New England Journal of Medicine, 1987,316(3):125-129.
[7]ROWE P S. Molecular biology of hypophosphataemic rickets and oncogenic osteomalacia[J]. Human Genetics, 1994,94(5):457-467.
[8]HANNA J D, NIIMI K, CHAN J C. X-linked hypophosphatemia. genetic and clinical correlates[J]. American Journal of Diseases of Children (1960), 1991,145(8):865-870.
[9]吴博. X连锁低血磷性佝偻病(XLH)成年患者的临床特征及磷酸盐尿性间叶组织肿瘤融合基因的研究[D]. 北京:北京协和医学院, 2016.
[10]LI S S, GU J M, YU W J, et al. Seven novel and six de novo PHEX gene mutations in patients with hypophosphatemic ri-
ckets[J]. International Journal of Molecular Medicine, 2016,38(6):1703-1714.
[11]GU J M, WANG C, ZHANG H, et al. Targeted resequencing of phosphorus metabolism-related genes in 86 patients with hypophosphatemic rickets/osteomalacia[J]. International Journal of Molecular Medicine, 2018,42(3):1603-1614.
[12]ZHANG C, ZHAO Z, SUN Y, et al. Clinical and genetic ana-
lysis in a large Chinese cohort of patients with X-linked hypophosphatemia[J]. Bone, 2019,121:212-220.
[13]GAUCHER C, WALRANT-DEBRAY O, NGUYEN T M, et al. PHEX analysis in 118 pedigrees reveals new genetic clues in hypophosphatemic rickets[J]. Human Genetics, 2009,125(4):401-411.
[14]SAITO T, NISHII Y, YASUDA T, et al. Familial hypophosphatemic rickets caused by a large deletion in PHEX gene[J]. European Journal of Endocrinology, 2009,161(4):647-651.
[15]LO F S, KUO M T, WANG C J, et al. Two novel PHEX mutations in Taiwanese patients with X-linked hypophosphatemic rickets[J]. Nephron Physiology, 2006,103(4):157-163.
[16]王青青,薛颖. 遗传性低磷血症性佝偻病简述[J]. 发育医学电子杂志, 2020,8(3):277-281.
[17]MKITIE O, DORIA A, KOOH S W, et al. Early treatment improves growth and biochemical and radiographic outcome in X-linked hypophosphatemic rickets[J]. The Journal of Clinical Endocrinology and Metabolism, 2003,88(8):3591-3597.
[18]YUE H, YU J B, HE J W, et al. Identification of two novel mutations in the PHEX gene in Chinese patients with hypophosphatemic rickets/osteomalacia[J]. PLoS One, 2014,9(5): e97830.
(本文編辑 黄建乡)
猜你喜欢
临床输血与检验(2022年3期)2022-06-22
中老年保健(2021年4期)2021-08-22
天津医科大学学报(2021年4期)2021-08-21
郑州大学学报(医学版)(2019年3期)2019-06-03
传染病信息(2019年2期)2019-05-17
中医眼耳鼻喉杂志(2019年2期)2019-04-13
农村青少年科学探究(2017年5期)2017-08-16
哈尔滨医药(2015年3期)2015-12-01
重庆医学(2015年12期)2015-03-05
疑难病杂志(2014年12期)2014-04-16
