
松果菊苷固体脂质纳米粒的制备及其在体肠吸收特性、体内药动学研究
2022-12-03 06:15决利利李晓婷王柯静周珊珊刘艳菊
中成药 2022年8期
决利利, 梁 婧, 李晓婷, 王柯静, 周珊珊, 刘艳菊*
(1.郑州工业应用技术学院医学院,河南 郑州 451150;2.无锡市农产品质量监测中心,江苏 无锡 214400)
松果菊苷是肉苁蓉、松果菊等中药的主要有效成分之一[1-2],具有抗肿瘤、降血脂、抗氧化、肝保护、神经保护、抗炎等活性,可用于多种系统疾病的治疗[1,3-5],但其溶解度仅为197.07 μg/mL,属于极微溶解药物,脂溶性差[6],而且体内容易代谢[4,7],稳定性不理想,导致口服吸收生物利用度低(0.83%)[5]。目前,鲜有关于松果菊苷新制剂技术的报道[6-7]。
固体脂质纳米粒是采用一种或几种脂质作为难溶性药物载体制成的实体骨架型纳米给药系统[8],可促进药物体外释放及肠道吸收,增加口服吸收生物利用度[9-11]。因此,本实验将松果菊苷制成固体脂质纳米粒,采用在体肠灌流方法考察该制剂在大鼠不同肠段中的吸收差异,并研究其体内药动学,以期为相关新制剂开发提供参考。
1 材料
1.1 仪器 安捷伦1200型高效液相色谱仪(美国安捷伦公司);MSE125P型分析天平(德国赛多利斯公司);JC-2LZF型旋转蒸发仪(青岛精诚仪器仪表有限公司);SZCL-2型磁力搅拌器(郑州生化仪器有限公司);BT100-1J/BZ15型蠕动泵(重庆科耐普蠕动泵有限公司);M-sizer 3000型粒度分析仪(英国马尔文仪器有限公司);JP-040PLUS型超声波仪(深圳市宝安区沙井洁盟德力生电器商行);MS-3型漩涡混合器(海门市其林贝尔仪器制造有限公司);HNDK200-2型氮气吹扫仪(上海琪摩电子科技有限公司)。
1.2 试剂与药物 松果菊苷(批号111670-201904,纯度98.3%)、绿原酸(批号110753-201716,纯度98.5%)对照品(中国食品药品检定研究院);松果菊苷原料药(批号190628,纯度95%,武汉卡诺斯科技有限公司)。K-R试液(批号20201025,北京百奥莱博科技有限公司);甘露醇(批号191123,四川博利恒药业有限公司);大豆磷脂(批号20190919,大连华农豆业科技发展有限公司);单硬脂酸甘油酯(批号20181208,上海吉至生化科技有限公司)。
1.3 动物 清洁级SD大鼠,雌雄不限,体质量(220±20)g,购自河南省动物实验中心,实验动物生产许可证号SCXK(豫)2016-0001。
2 方法与结果
2.1 松果菊苷含量测定
2.1.1 色谱条件 Venusil XBP-C18色谱柱(150 mm×4.6 mm,5 μm);Phenomenex C18预柱(4 mm×2 mm,5 μm);流动相乙腈-0.4%磷酸(60∶40);体积流量1.0 mL/min;柱温30 ℃;检测波长333 nm。
2.1.2 供试品溶液制备 取1 mL固体脂质纳米粒混悬液至50 mL量瓶中,加入10 mL甲醇超声提取5 min,静置20 min,流动相定容至刻度,摇匀,过0.45 μm微孔滤膜,即得。
2.1.3 线性关系考察 取20.20 mg对照品至100 mL量瓶中(减重法称量),乙腈定容至刻度,得202 μg/mL贮备液,密封后置于冰箱中保存,取适量,流动相稀释至20.2、10.1、2.02、1.01、0.505、0.050 5 μg/mL,即得对照品溶液,在“2.1.1”项色谱条件下进样测定。以对照品质量浓度为横坐标(X),峰面积为纵坐标(Y)进行回归,得方程为Y=17.215 3X-0.419 4(r=0.999 9),在0.050 5~20.2 μg/mL范围内线性关系良好。
2.1.4 方法学考察 取“2.1.2”项下供试品溶液,于0、3、6、9、12、24 h在“2.1.1”项色谱条件下进样测定,测得松果菊苷含量RSD为1.06%,表明溶液在24 h内稳定性良好。按“2.1.2”项下方法平行制备6份供试品溶液,在“2.1.1”项色谱条件下进样测定,测得松果菊苷含量RSD为1.67%,表明该方法重复性良好。取0.050 5、2.02、20.2 μg/mL对照品溶液,在“2.1.1”项色谱条件下进样测定,测得松果菊苷含量RSD分别为1.14%、0.38%、0.49%,表明仪器精密度良好。取9份固体脂质纳米粒混悬液,每份0.5 mL,置于9个50 mL量瓶中,分为3组,分别加入202 μg/mL贮备液0.5、1、1.5 mL,按“2.1.2”项下方法制备供试品溶液,在“2.1.1”项色谱条件下进样测定,测得松果菊苷平均加样回收率分别为101.72%、99.46%、100.85%,RSD分别为0.62%、1.17%、1.92%。
2.2 固体脂质纳米粒制备 采用冷均质法制备[8,12]。取0.8 g单硬酯酸甘油酯、0.4 g大豆磷脂,65 ℃水浴加热熔融,加入50 mg松果菊苷,磁力搅拌溶解得熔融液,迅速置于-65 ℃超低温冰箱中12 h得固体物质,适当粉碎后置于120 mL 1%泊洛沙姆188溶液(4 ℃)中,胶体磨中形成粗分散体系,在压力60 MPa条件下循环均质8次,过0.45 μm微孔滤膜,加蒸馏水至120 mL,即得。再以5%甘露醇为冻干保护剂,加到固体脂质纳米粒混悬液中,摇匀,在-30 ℃下预冻2 d,迅速置于冻干机中,抽真空后冷冻干燥1 d,得到冻干粉。
2.3 包封率、载药量测定 取固体脂质纳米粒混悬液1 mL,按“2.1.2”项下方法制备供试品溶液,测定松果菊苷总量W0;取1 mL固体脂质纳米粒混悬液至离心管中,4 ℃、12 500 r/min离心30 min,取上清液,测定游离松果菊苷量W1;将沉淀物在-30 ℃下预冻2 d,迅速置于冻干机中,抽真空后冷冻干燥1 d,测定松果菊苷和脂质总量W,计算包封率、载药量,公式分别为包封率=[(W0-W1)/W0]×100%、载药量=[(W0-W1)/W]×100%。结果,3批固体脂质纳米粒混悬液平均包封率为(83.34±1.41)%,载药量为(3.19±0.23)%。
2.4 粒径、Zeta电位测定 取固体脂质纳米粒混悬液0.1 mL,蒸馏水稀释40倍后测定其粒径、Zeta电位。结果,固体脂质纳米粒混悬液平均粒径为(192.07±9.14)nm,PDI为0.107±0.013,而其冻干粉复溶后两者分别为(237.77±9.14)nm、0.186±0.024,见图1;两者Zeta电位分别为(-22.64±2.13)、(-18.71±2.24)mV,见图2。
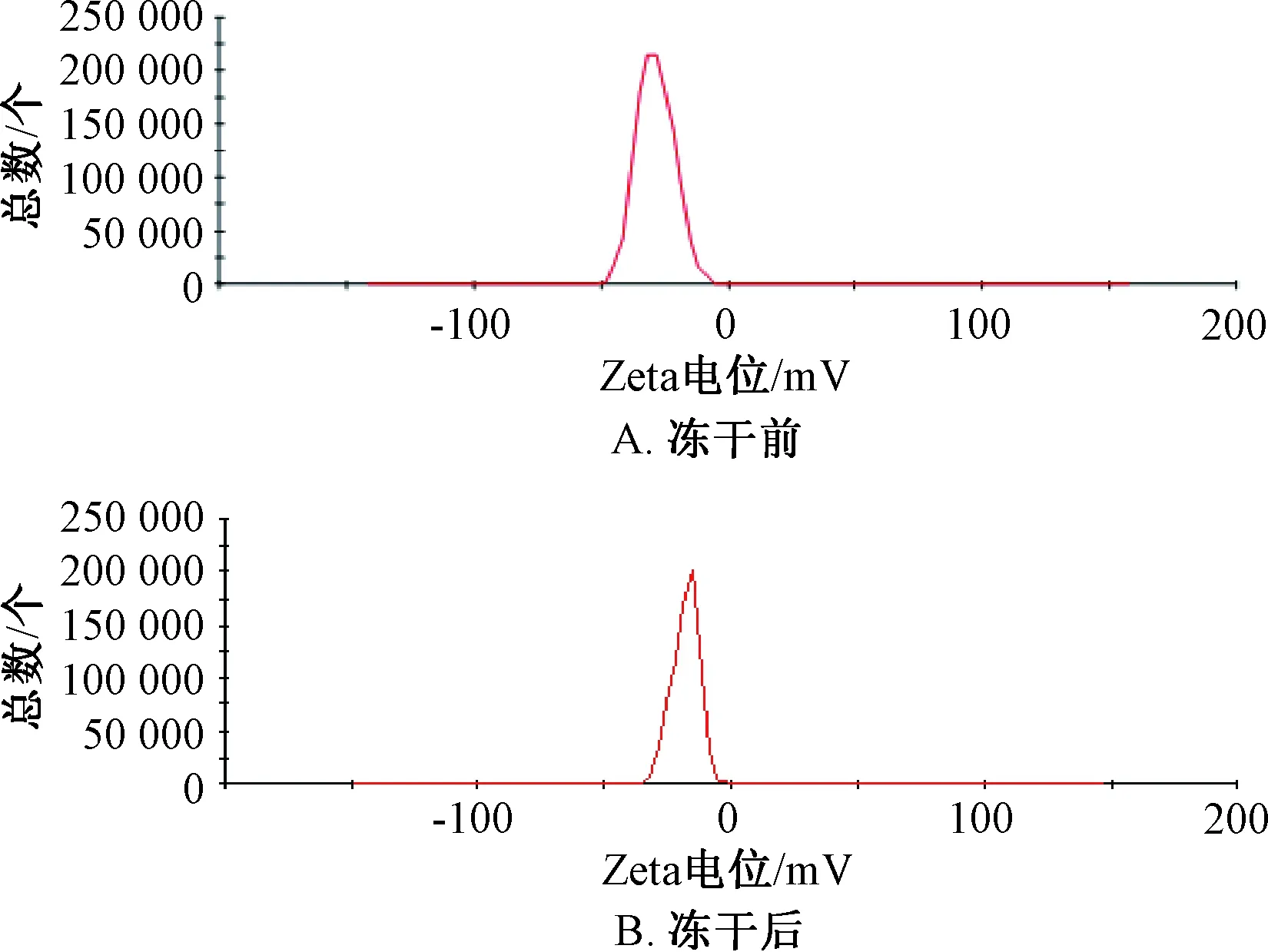
图2 松果菊苷固体脂质纳米粒Zeta电位
2.5 XRPD扫描 取松果菊苷、空白辅料、物理混合物(松果菊苷与空白辅料比例同固体脂质纳米粒冻干粉)、固体脂质纳米粒冻干粉适量,设定扫描范围为3°~45°,速度为6°/min,结果见图3。由此可知,松果菊苷在10°~30°范围内出现多处晶型峰;物理混合物在11.9°、16.3°、16.8°、25.8°等处仍可见其晶型峰;固体脂质纳米粒冻干粉仅见空白辅料的晶型峰,松果菊苷晶型峰均消失,表明该成分变为无定型物质。
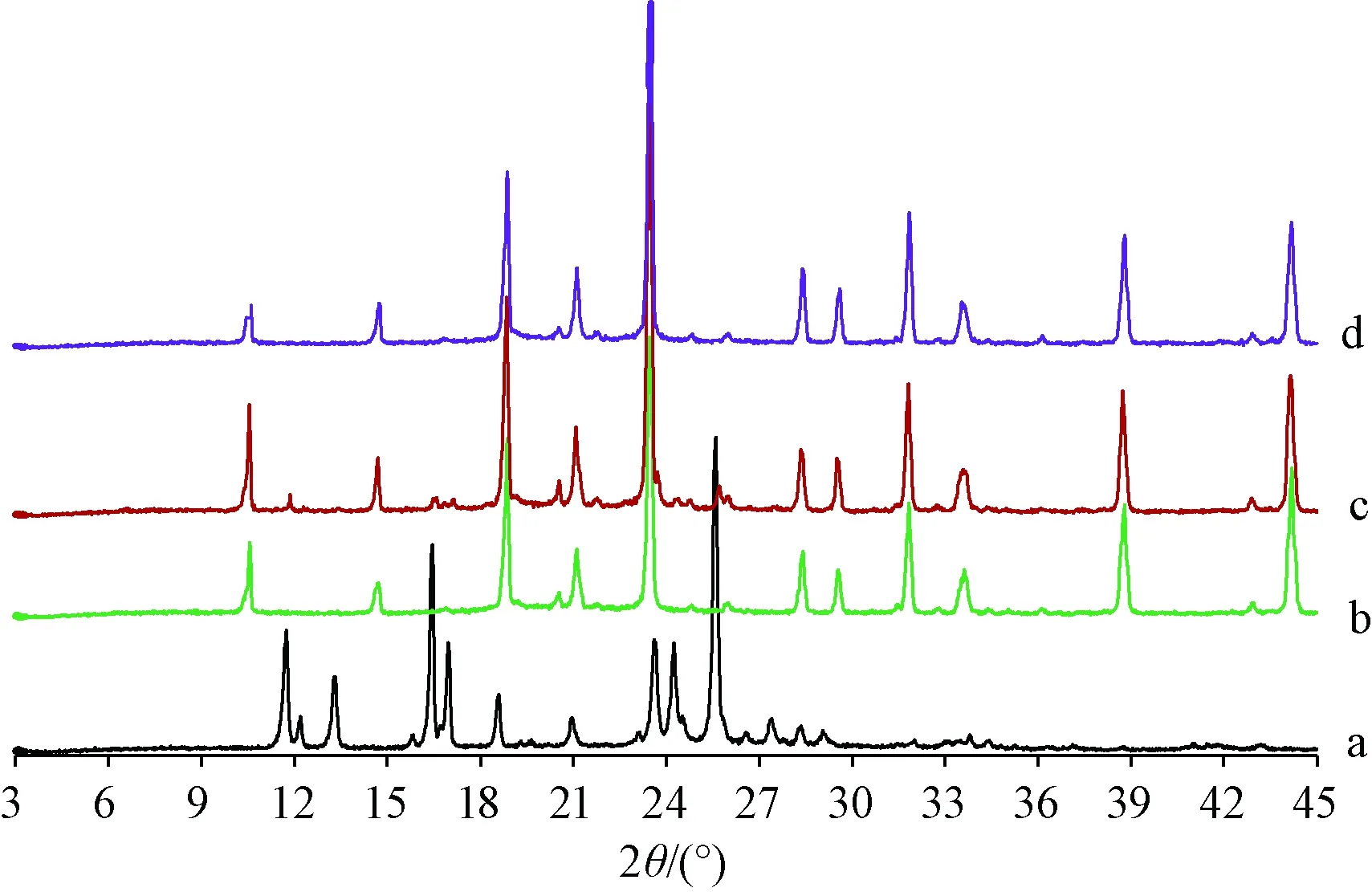
注:a~d分别为松果菊苷、空白辅料、物理混合物、固体脂质纳米粒冻干粉。
2.6 油水分配系数测定 参考文献[13-14]报道,取过量松果菊苷、物理混合物、固体脂质纳米粒冻干粉适量,加入正辛醇饱和的水相,超声处理30 min,固定于振荡器上,37 ℃、100 r/min振荡1 d,取上层混悬液,过0.22 μm水膜,测定在水中的溶解度C1,同法测定在蒸馏水饱和正辛醇中的溶解度C2,计算油水分配系数P,公式为P=(C1-C2)/C2,并计算lgP。结果,松果菊苷、物理混合物、固体脂质纳米粒冻干粉lgP分别为0.106、0.139、0.807,表明固体脂质纳米粒可增加松果菊苷油水分配系数,有助于该成分透膜吸收。
2.7 体外释药研究 取适量松果菊苷及其固体脂质纳米粒冻干粉(含30 mg松果菊苷),加入3 mL空白释放介质得混悬液,转移至透析袋中(截留分子量12~14 kDa),一端用细尼龙绳扎紧,另一端固定于搅拌桨上,释药介质为1 000 mL蒸馏水,设定转速、温度分别为75 r/min、37 ℃,于0、0.5、0.75、1、1.5、2、3、4、6、8、12、18、24、36 h各取样3 mL,补加空白释放介质,过0.45 μm微孔滤膜,测定松果菊苷含量,计算累积释放度,结果见图4。由此可知,在8 h,原料药释放度达94.48%,而固体脂质纳米粒仅为65.04%,属于快速释药期;在8~36 h,固体脂质纳米粒累积释放度呈缓慢增加趋势,36 h内达84.06%。
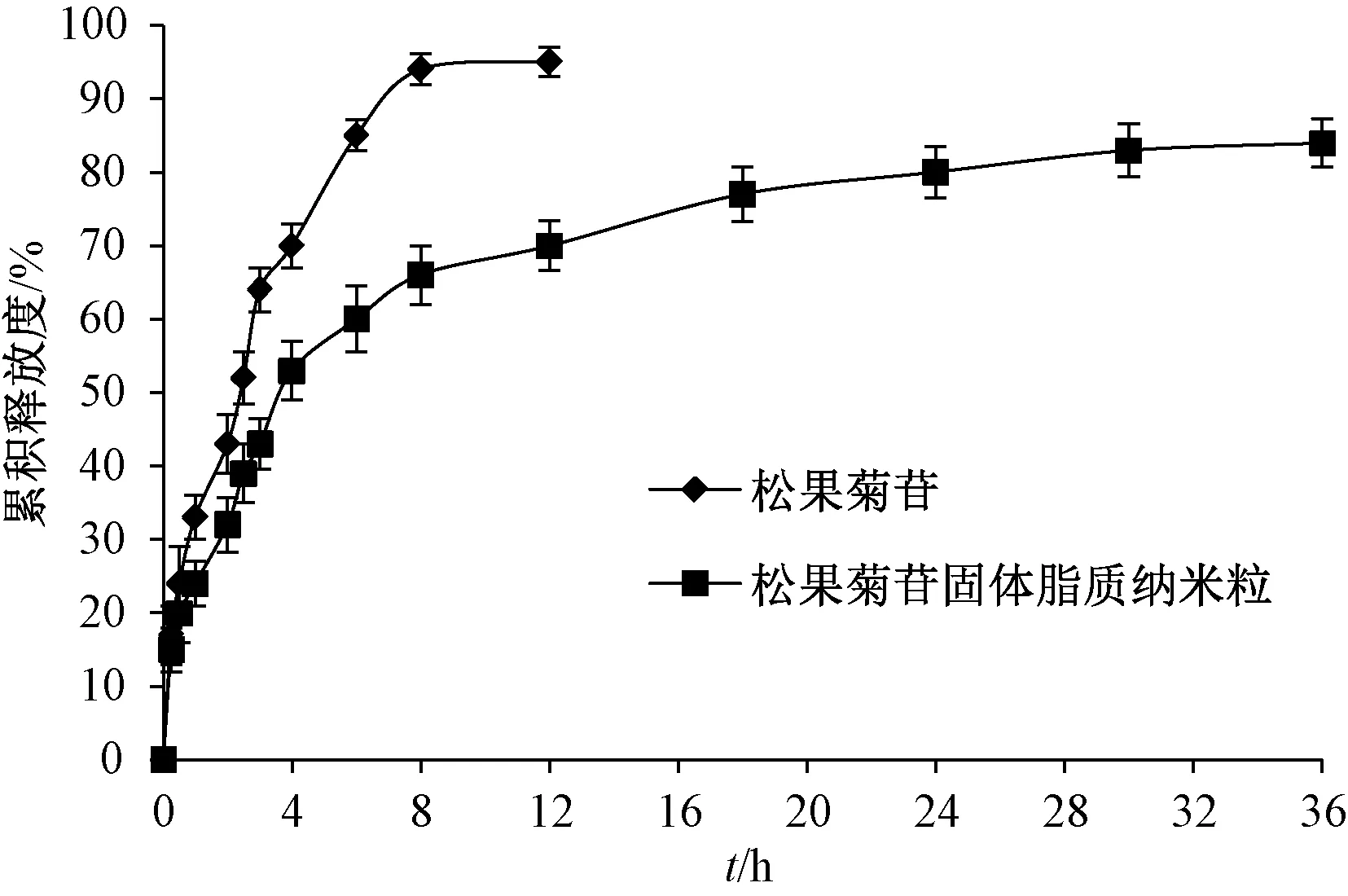
图4 松果菊苷体外释药曲线(n=6)
2.8 在体肠吸收特性研究
2.8.1 肠灌流溶液制备 取K-R试液进行在体肠灌流操作,收集流出液,作为空白肠灌流液。取松果菊苷及其固体脂质纳米粒冻干粉适量,空白肠灌流液配制15 mL(以松果菊苷计,质量浓度为40 μg/mL),即得。
2.8.2 含药灌流液稳定性 取“2.8.1”项下含药灌流液,置于37 ℃恒温水浴中,于0、15、30、45、60、90、120 min各取样1 mL,过0.45 μm微孔滤膜,在“2.1.1”项色谱条件下进样测定。结果,原料药、固体脂质纳米粒灌流液峰面积RSD分别为0.71%、1.14%,表明含药灌流液在K-R试液中稳定性良好。
2.8.3 肠壁对灌流液中药物的物理吸附 剪取肠段,K-R试液冲洗3次,置于松果菊苷及其固体脂质纳米粒灌流液中孵育2 h,K-R试液冲洗2次,分别测定孵育前和孵育2 h后松果菊苷剩余药量。结果,原料药、固体脂质纳米粒灌流液剩余药量分别为98.74%、101.26%,RSD分别为0.62%、0.81%,表明大鼠肠壁对灌流液中药物无明显的吸附作用。
2.8.4 实验操作 取禁食12 h(可自由饮水)的大鼠12只,随机分为松果菊苷组、松果菊苷固体脂质纳米粒组,腹腔注射戊巴比妥钠(50 mg/kg)麻醉,仰卧固定于37 ℃保温垫上,实验用管道均在同浓度灌流液中过夜浸泡,沿腹部中线打开腹腔,分离十二指肠、空肠、回肠、结肠,在待灌流肠段上部和下部用手术剪开口(勿剪断),插管后扎紧,K-R试液冲洗内容物。大鼠腹部覆盖浸有生理盐水的纱布(适时洒生理盐水以保湿),设置灌流液体积流量为0.2 mL/min,开启蠕动泵,收集流出液,1 h后停止灌流,K-R试液冲洗2次,合并流出液,采用重量法对其体积进行校正。剪断灌流结束肠段,记录内径(r)、长度(l),流出液6 500 r/min离心30 min,测定松果菊苷质量浓度,计算吸收速率常数Ka、表观吸收系数Papp,公式分别为Ka=(1-CoutQout/CinQin)Q/V、Papp=-Qln(CoutQout/CinQin)/2πrl,其中Qin、Qout分别为进、出灌流液体积,Cin、Cout分别为进、出灌流液浓度,Q为灌流液体积流量,V为待考察肠段体积。
2.8.5 结果分析 表1显示,松果菊苷在各肠段中均有一定程度的吸收,其中结肠段吸收较差;将该成分制成固体脂质纳米粒后,Ka、Papp在各肠段中均升高(P<0.01),其中结肠段更明显,推测可能存在结肠靶向。

表1 松果菊苷吸收参数
2.9 体内药动学研究
2.9.1 血浆处理 参考文献[2]报道,取血浆100 μL至5 mL离心管中,加入50 μL内标(绿原酸)溶液(10 μg/mL),涡旋15 s后加入1 mL甲醇沉淀蛋白,密封后涡旋3 min,8 500 r/min离心6 min,移取上层有机相至另一离心管中,置于40 ℃氮吹仪中缓慢吹干得残渣,加入100 μL乙腈超声处理30 s进行复溶,进样分析。
2.9.2 分组、给药与采血 取松果菊苷适量,加入3 mL 0.5%CMC-Na制成混悬液(6 mg/mL),同法制备固体脂质纳米粒冻干粉混悬液。取禁食12 h的大鼠12只(雌雄兼具),分为2组,灌胃给予2种混悬液(50 mg/kg),于0.25、0.5、1、1.5、2、3、4、6、8、12 h采血约0.3 mL,置于涂抹肝素的离心管中,振荡混匀,3 000 r/min离心3 min,保存于-15 ℃冰箱中。
2.9.3 线性关系考察 大鼠麻醉后心脏采血,置于涂抹肝素的离心管中,振荡混匀,3 000 r/min离心3 min,取上清液,即为空白血浆。取松果菊苷对照品适量,空白血浆制成1 600、800、400、200、100、20 ng/mL血浆对照品溶液,按“2.9.1”项下方法处理,在“2.1.1”项色谱条件下进样测定。以对照品质量浓度为横坐标(X),峰面积为纵坐标(Y)进行回归,得方程为Y=0.016 2X+0.110 7(r=0.996 3),在20~1 600 ng/mL范围内线性关系良好。
2.9.4 方法学考察 取给药固体脂质纳米粒后1 h的血浆,于0、2、4、6、8、12 h在“2.1.1”项色谱条件下进样测定,测得松果菊苷含量RSD为8.61%,表明血浆在12 h内稳定性良好。取20、400、1 600 ng/mL血浆对照品溶液适量,在“2.1.1”项色谱条件下进样测定6次,测得松果菊苷含量RSD分别为9.36%、5.19%、6.04%,表明仪器精密度良好。取上述3种溶液适量,按“2.9.1”项下方法处理,在“2.1.1”项色谱条件下进样测定,测得松果菊苷含量RSD分别为10.17%、7.56%、8.90%,表明该方法重复性良好。取上述3种溶液适量,在“2.1.1”项色谱条件下进样测定,测得松果菊苷平均加样回收率分别为90.14%、95.62%、92.38%,RSD分别为7.16%、4.31%、5.28%。
2.9.5 结果分析 图5、表2显示,与原料药比较,固体脂质纳米粒tmax延长(P<0.01),Cmax、AUC0~t、AUC0~∞升高(P<0.01),相对生物利用度提高至3.82倍。
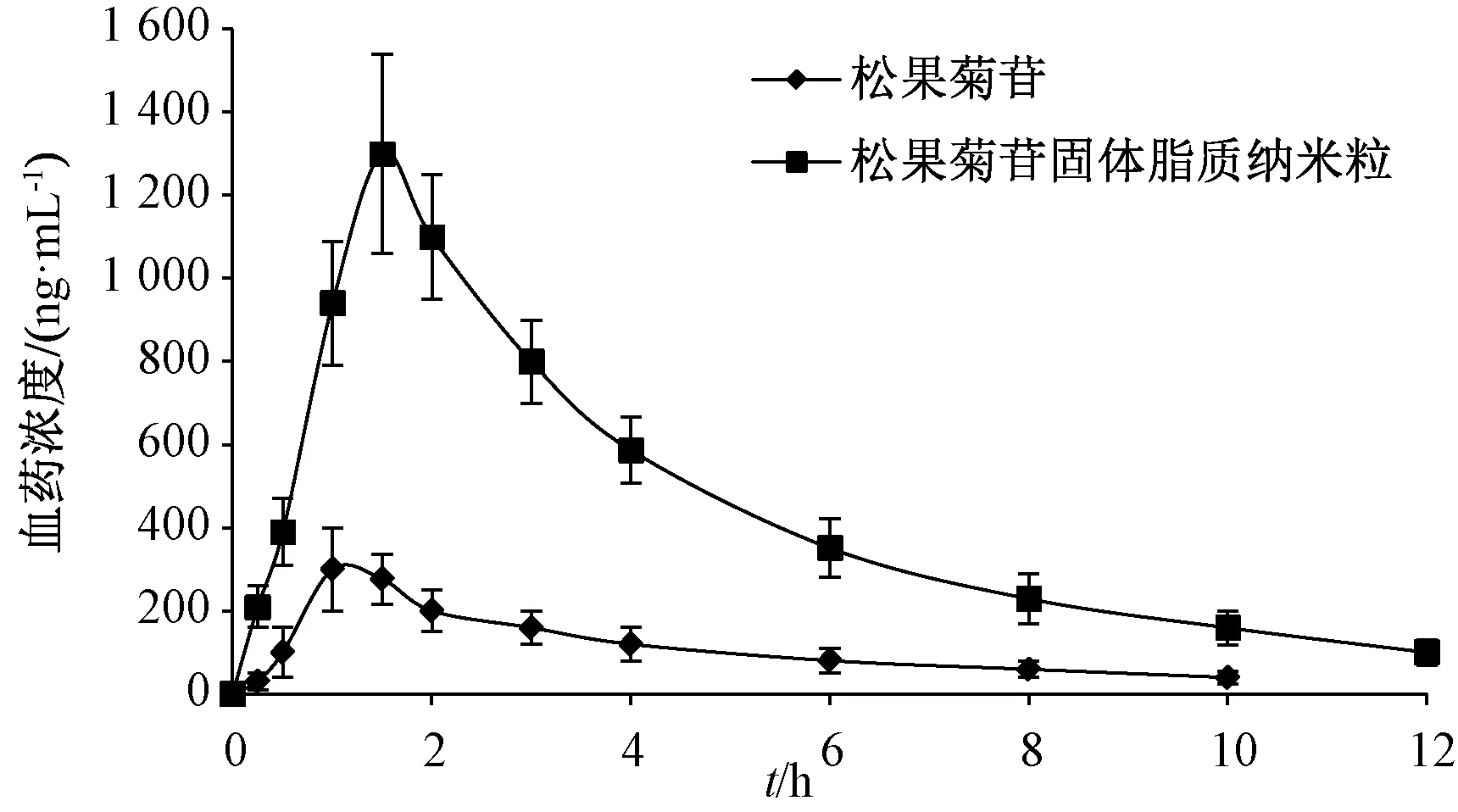
图5 松果菊苷血药浓度-时间曲线(n=6)

表2 松果菊苷主要药动学参数
3 讨论
陈静等[15]采用乳化固化法制备松果菊苷固体脂质纳米粒,但其包封率不足60%,可能是药物处方中含有表面活性剂,在超声增溶作用下使药物进入水相,从而影响脂质载体对药物的包裹效率。本实验采用冷均质法制备该制剂,无需超声提取,有助于减少药物向水相的转移[8],包封率达(80.24±1.53)%。
由于在体肠灌流实验时大鼠各个肠段除了吸收药物外,同时还吸收水分,故本实验采用重量法对灌流体积进行校正,以保证实验结果的准确性。结果,松果菊苷固体脂质纳米粒Ka、Papp在大鼠结肠中的升高程度最大,可能是该处存在大量淋巴液[16],从而增加该成分淋巴转运所致。
一般认为,当Papp小于3×10-6时,药物吸收较差[17],而松果菊苷各肠段Papp均小于该数值,提示肠道吸收受限是该成分口服吸收较差的原因之一,而本实验将其制成固体脂质纳米粒,可显著提高该参数。本实验发现,松果菊苷在固体脂质纳米粒中变为无定型物质,该形态往往比晶型药物具有更高的生物利用度[18-19];固体脂质纳米粒提高了松果菊苷油水分配系数,也有助于促进该成分吸收[14,20],从而提高其口服吸收生物利用度;与原料药比较,固体脂质纳米粒tmax延长,可能是因为脂质载体的包裹作用及胃肠道对纳米药物的吸附作用,导致部分药物释放延缓,从而影响tmax。
猜你喜欢
护理与康复(2021年4期)2021-12-07
保健医苑(2021年4期)2021-12-01
载人航天(2021年5期)2021-11-20
现代仪器与医疗(2021年1期)2021-06-09
健康之家(2021年19期)2021-05-23
中国食品(2020年23期)2020-12-23
智慧健康(2020年5期)2020-03-24
中国中医药信息杂志(2019年10期)2019-11-15
家庭百事通·健康一点通(2019年8期)2019-08-29
医学信息(2015年5期)2015-03-31
