
单原子催化剂Ir1/MoS2表面上的NH3吸附与直接分解的第一性原理
2022-12-07 09:36肖香珍银召利张建伟
原子与分子物理学报 2022年1期
肖香珍, 银召利, 张建伟
(1. 河南科技学院 实验管理中心, 新乡 453003; 2. 河南科技学院 新科学院, 新乡 453003)
1 引 言
由于过度金属结构上的特殊性,金属单晶表面常常被用来作为吸附或催化反应的催化剂,它们在氮氧化物的消除、氨气的解离等方面有着独特的催化性能,因此受到了极大关注[1-4].近几年贵金属Ir作为一个有潜力的多相反应催化剂,在表面科学领域的应用比较广泛,特别对于NH3的解离与氧化表现出较好的低温催化活性及产物选择性.但在催化反应中纯金属催化剂的金属利用效率较低[5-7],起催化作用的只有极少数金属活性组分,催化成本较高.最近研究发现单原子催化剂可以实现原子利用率的最大化,且有着高活性、高稳定性和高选择性,因此吸引了众多研究者的关注[8-14].
随着量子化学理论、高性能计算技术的飞速发展,理论计算应经成为催化科学实验研究的重要组成部分.为揭示单原子与载体之间的相互作用和单原子催化剂的催化性能, 研究者们已经开展了大量关于单原子催化剂的密度泛函理论(DFT)计算,例如模拟计算单原子可能的吸附位点[8]、反应气体的真实吸附、催化反应路径及能垒大小等[15, 16].目前单原子催化剂选用的载体是金属、金属氧化物及其二维材料[17-20].在众多二维材料中,单层MoS2因在光致发光、光吸收、和催化方面等表现出优良的特性受到广泛关注.
然而,Ir原子附着在单原子层MoS2上气体NH3的吸附与解离至今未见报道.本论文是在先前研究纯贵金属Ir催化剂表面NH3吸附与解离的工作基础上[21-24],选择单层二维材料MoS2作为载体,单个贵金属原子Ir吸附在MoS2上形成单原子催化剂Ir1/MoS2,采用密度泛函方法计算了NH3在单原子催化剂Ir1/MoS2上的最优吸附位置以及直接分解情况.本研究目的是既能通过NH3催化分解得到高纯度的氢,又能减少贵金属的使用量,实现贵金属原子利用率的最大化.
2 计算模型和方法
MoS2的晶体结构类似于石墨烯[25],具有二维层状结构,即Mo 原子在中间,上下分别由共价键连接着S原子,形成三明治式S Mo S的结构.
本文计算均采用3×3 单层MoS2超胞Slab模型,所建超胞模型的俯视图和侧视图如图1所示,由9个Mo原子和18个S 原子组成.优化后得到的晶胞参数0.319 nm与实验值0.316 nm[26]吻合很好.所有计算均采用DFT方法结合周期性平板模型进行结构优化[27, 28],电子结构计算均由 VASP[29, 30]软件包完成.交换-关联泛函采用GGA-PW91来计算[31],电子与离子实间的相互作用采用PAW方法[32]描述.平面波基组的截断能均为500 eV.表面布里渊区K点网格密度取为5×5×1[33].结构优化时固定下面两层Mo和S原子,吸附物Ir和N2以及顶层S原子位置S随吸附物弛豫.真空层厚度取为1.5 nm.所有优化得到的最优构型都通过频率分析加以验证.

图1 单层MoS2超胞的俯视图(a)和侧视图(b)及单原子催化剂Ir1/MoS2(c),其中,黄色为S原子,紫色为Mo原子,深黄色为Ir原子.
本文采用vasp软件包的improved dimer method(IDM)[34, 35]方法搜索反应过渡态、确定最小能量路径以及活化能,对过渡态进行频率验证,即过渡态有且只有一个虚频.
3 结果与讨论
3.1 单原子Ir的落位
通过计算和比较不同的吸附位点的吸附能来确定体系Ir1/MoS2的最优构型.MoS2的三个原子层按照平面六角阵列排列,其有两个典型考查位点:四重空位(fcc位)和三重空位(hcp位),因此计算了Ir原子在两个吸附位点的吸附能.
吸附能可以反映吸附前后各物质总能量的变化[36], 将MoS2视为吸附表面,吸附能依下式计算:
Eads=EIr+EMoS2-EIr/MoS2
(1)
计算得到fcc位的吸附能为:Eads=2.98 eV,hcp位的Eads=3.98 eV.hcp位吸附能较大,吸附稳定,且通过频率分析显示hcp位对应无虚频.其结构俯视图示于图1(c).
3.2 NH3在体系Ir1/MoS2上吸附
以NH3为探针分子,考察其在单原子Ir上的不同吸附构型,如图2(a)、(b)、(c)、(d)所示.此时吸附能定义为:

图2 NH3在体系 Ir1/MoS2上吸附的构型图
Eads=ENH3+EIr/MoS2-ENH3/Ir/MoS2
(2)
不同吸附构型均是NH3分子通过N原子和MoS2表面上的Ir原子相连.图2(a)和图2(c)结构相似,NH3的C3轴垂直于体系Ir1/MoS2,不同的是(a)结构中N-H键投影至 Ir -S键方向,(c)中三个N-H键是投影至六角表面的fcc位.(a)和(c)两种构型的吸附能相同,均为1.58 eV,见表1.Ir-N和N-H键长(dIr-N(nm) 、dN-H(nm) )以及夹角∠HNH差异很小,与气相NH3的键长(dN-H=0.102 nm)和夹角(∠HNH=107.3°)比较,基本保持了气态分子的分子结构特征,可见吸附前后分子结构没有发生明显改变.
图2 (b)和(d)均为倾斜结构,但倾斜程度不同.(b)构型中倾斜方向上N、Ir、S原子之间的两个夹角∠NIrS分别为100.4°、97.9°,但(d)构型倾斜方向上两个∠NIrS分别为108°、100.7°.计算了比较了两种构型的吸附能,如表1,(d)的结构相对稳定,这个最稳定吸附位类似于NH3在纯金属Ir(110)上吸附结构[23],而NH3在Ir(110)面上的吸附能仅为1.11 eV,说明NH3在单原子催化剂上更易吸附.(d)结构中吸附后NH3的夹角∠HNH变化较大,说明吸附过程中受到原子Ir某些轨道的扰动.

表1 NH3在Ir1/MoS2体系上吸附不同构型的几何参数
3.3 NHX(X=0-3)+(3-X)H等在Ir1/MoS2体系上的共吸附

EA+B/S-ads=EA+B+Esurf-EA+B/surf
(3)
(4)
其中EA+B、EA、EB、Esurf、EA+B/surf分别表示A+B吸附分子、B吸附分子的能量和A+B共吸附在体系Ir1/MoS2表面的能量.相互作用能被定义为共吸附结构的吸附能与单独最稳定吸附时的吸附能之差.


表2 各共吸附体系的吸附能


图3 NHx(x=0-3)+(3-x)H共吸附在体系Ir1/MoS2上的吸附能随H原子个数的变化
3.4 NH3在体系Ir1/MoS2上的直接脱氢
本文中按照逐步脱氢的反应机理,以各吸附体系的最稳定共吸附结构为反应物或生成物,在经历反应时,旧键断裂,新键形成,本研究包括以下四步基元反应:

对于NH3的第一步脱氢反应,以其吸附在Ir原子的倾斜结构作为反应物,如图4(a)所示,此过程中,其中一个N-H键的键长逐渐拉长,当拉长到0.151 nm,形成过渡态,如图4(b)所示.此后,N-H键逐渐断裂,H原子移动到距离N原子0.238 nm,倾斜吸附在Ir原子上,如图4(c),过渡态的虚频值为1051.6 cm-1,对应于相应的N-H键的伸缩振动.NH2倾斜吸附在Ir原子上,在原NH3倾斜吸附的另一方.该过程中的活化能为1.41 eV,反应热为0.11 eV,见表4,此反应为吸热反应.
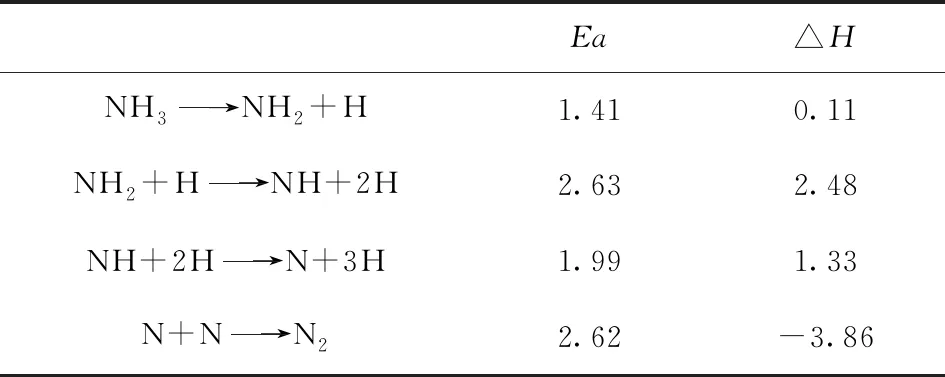
表4 在Ir1/MoS2体系上NH3分解和N2重组的活化能及反应热(单位:eV)
在第二步脱氢反应中,以第一步的终态为反应物,在反应过程中,NH2的一个N-H键由始态的0.102 nm被拉伸到0.194 nm,达到过渡态,此时N-H键基本完全断开,如图4(d),过渡态的虚频为1017.8 cm-1.被拉伸的H原子被最近的S原子吸附,剩下的N-H停留在Ir原子上,此时即为反应的终态,如图4(e)所示.此步反应的能垒为2.63 eV,反应热为2.48 eV,为吸热反应.
第三步反应过程中,以图4(e)结构为反应物,图4(g)为产物结构,该反应过程中,当N-H键拉长到0.152 nm,形成了过渡态(TS),过渡态的虚频为1065 cm-1,如图4(f)所示.此后N原子不移动,H原子继续拉伸到离N原子0.246 nm的位置,N、H仍均吸附在Ir原子上.该反应的活化能为1.99 eV,反应热为1.33 eV,说明该反应为吸热反应.
当两个N原子共吸附在Ir1/MoS2体系上时,通过优化结构得到当两个N原子均吸附在Ir原子上,吸附最稳定,此时N-N之间的距离为0.290 nm,N1-Ir和N2-Ir键长分别为0.173 nm、0.171 nm,如图5.将两个N原子共吸附在Ir1/MoS2上当作直接形成N-N键的初态,在TS中,N-N之间的距离缩短为0.222 nm,过渡态的虚频为216.1 cm-1,对应N-N键的伸缩振动,其中N2-Ir键已伸长至0.194 nm,表明此时N2-Ir键强度减弱,但要形成垂直结构的N2分子,N2原子还要翻转至N1原子上方,此时需要越过较高能垒才能重组形成N2,此时N-N键长为0.114 nm,能垒为2.62 eV,整个过程体系放热3.86 eV.

图4 NH3脱氢反应相应的始态,过渡态和终态的结构示意图,以及势能面图
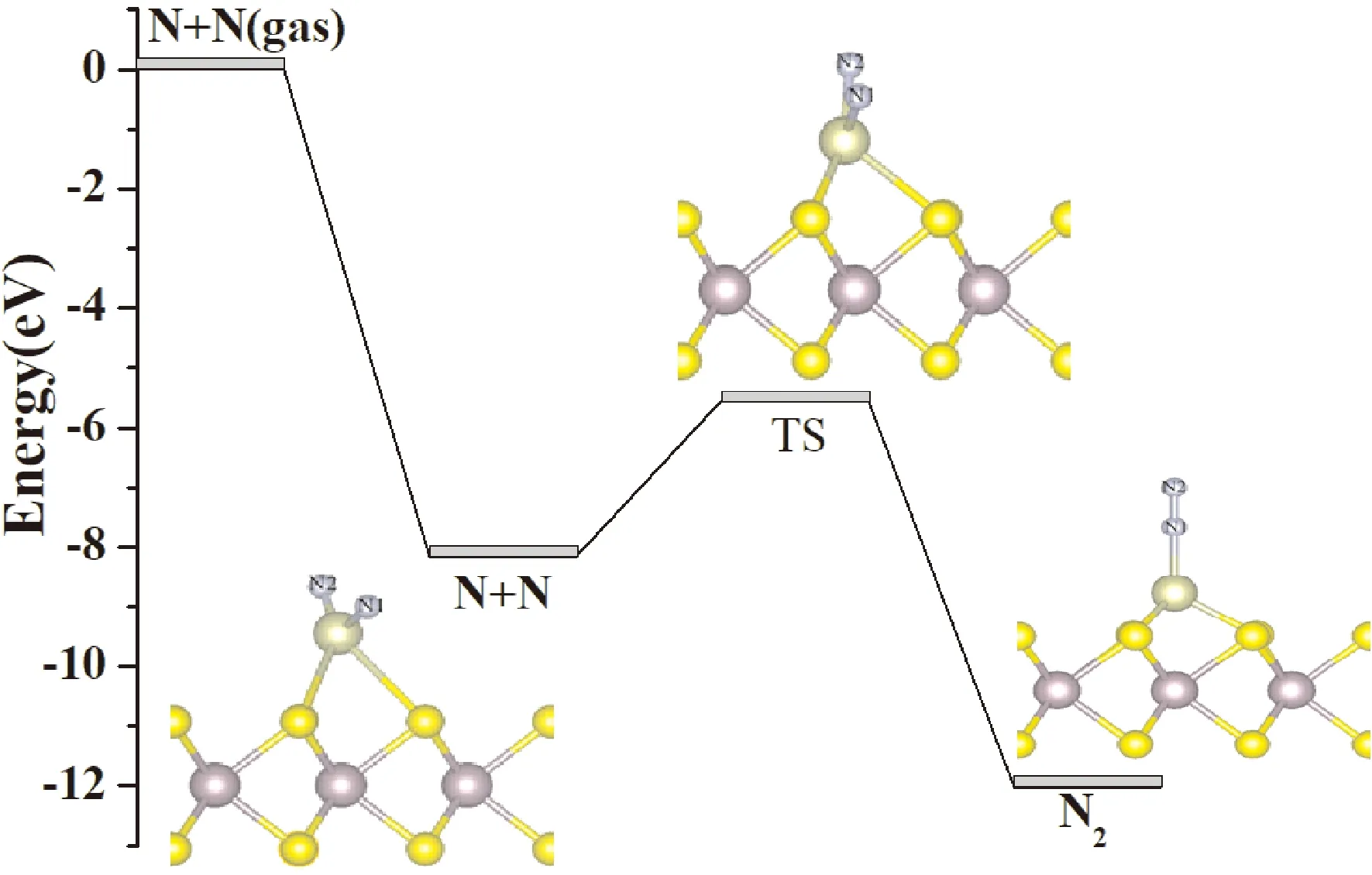
图5 N-N聚合反应的势能面图,以及相应的始态,过渡态和终态的结构示意图
为了更好的了解NH3解离过程,在图4(h)中列出了反应的势能面.从图表中显示,NH3在该体系上的吸附能大于第一步脱氢所需要的活化能,且整个反应中,此步反应的能垒最小,0说明NH3在Ir1/MoS2上不仅能够分解,而且较容易断裂N-H键形成NH2和H.而反应NH2+H→NH+2H的能垒高于其他几步反应,说明NH2的N-H键很难断裂,在Ir1/MoS2体系上吸附比较稳定,是整个反应过程中最主要的吸附物,此步反应也是整个分解反应的速控步骤.两个N原子共吸附后需克服2.62 eV的能垒才能重组形成产物N2,此能垒接近第二步脱氢反应的活化能.
通过对三步脱氢反应的初态、过渡态以及终态的N-Ir键长的比较,如下图6所示.发现随着反应的进行,NHx物种上的H原子个数减少,而共吸附的H原子数增多,N原子到单原子金属Ir的距离变小[37],表明N-Ir成键作用增强,吸附越稳定,上一节也显示共吸附时的吸附能逐渐增大.

图6 脱氢反应过程中Ir原子与N原子之间距离的变化
4 结 论
利用第一性原理并结合平板模型,对NH3在单原子催化剂Ir1/MoS2上的吸附与脱氢解离机理进行了研究.结果表明,单个Ir原子附着在MoS2表面的三重空位hcp位时吸附能为3.98 eV,此时整个体系稳定. NH3分子在体系Ir1/MoS2上是通过N原子和MoS2表面上的Ir原子相连,它的C3垂直于体系Ir1/MoS2,三个N-H键指向远离表面的方向,且投影至 Ir -S键方向,吸附能为1.63 eV.在整个NH3的解离反应中,NH3的第一步脱氢的活化能是整个反应中最小的,说明NH3的N-H键相对容易断裂.第二步NH2+H→NH+2H反应中,能垒最高,NH2的N-H键就较难断裂.随着反应的进行,共吸附的H原子增多,NHx物种的H原子个数减少,N原子到金属Ir表面的距离也变小,N-Ir成键作用增强,吸附能逐渐增大.
猜你喜欢
四川大学学报(自然科学版)(2022年4期)2022-07-22
北京航空航天大学学报(2022年5期)2022-06-06
华东理工大学学报(自然科学版)(2022年2期)2022-04-29
浙江大学学报(理学版)(2021年6期)2021-12-02
中学课程辅导·教学研究(2021年8期)2021-07-14
化学工业与工程(2021年3期)2021-06-25
青岛大学学报(工程技术版)(2019年2期)2019-09-10
电脑知识与技术(2018年3期)2018-03-21
哈尔滨理工大学学报(2017年1期)2017-04-08
科技视界(2016年24期)2016-10-11
