一例3M 综合征合并Netherton 综合征家系的遗传学研究
2022-12-23 13:13黎伟豪黄秀静叶燕绸
分子诊断与治疗杂志 2022年11期
黎伟豪 黄秀静 叶燕绸★
3M 综 合 征(Three M syndrome,OMIM #273750)是一种常染色体隐性遗传病,由Miller、Mukusick、Malvaux 三位遗传学家于1975 年首次发现并报道,其主要症状为严重宫内和出生后生长迟缓,特殊面容,骨骼发育异常[1]。已报道3 个致病基因:CUL7(MIM 609577)基因、OBSL1(MIM 610991)基因和CCDC8(MIM 614145)基因,分别引起3M 综合征的Ⅰ型、Ⅱ型、Ⅲ型三种类型。CUL7基因变异导致3M 综合征占77.5%,OBSL1基因变异占16%,CCDC8基因变异少于5%,其他不明确基因变异约占1.5%[2-3]。
Netherton 综 合 征(Netherton syndrome,NS,OMIM#256500)也是一种常染色体隐性遗传病,于1958 年由Earl Netherton 首次报道,由SPINK5(MIM 605010)基因变异引起。NS 典型的临床表现包括先天性鱼鳞病样红皮病/回旋形线状鱼鳞病、毛发异常(套叠性脆发)和特应性过敏体质以及生长发育迟缓等[4]。NS临床表现与其他多种皮肤病相似,可能造成漏诊,估计NS约占新生儿红皮病的20%[5]。
本文对一例产前超声影像提示股骨发育速度明显缓慢、面部发育异常、胸骨内陷的胎儿进行了家系全外显子组测序(Whole Exome Sequencing ,WES),研究报告如下:
1 材料与方法
1.1 一般资料
孕妇27 周岁,孕2 产1,早孕期超声影像检查提示胎儿颈后透明带(Nuchal Translucency,NT)1.7 mm;早、中孕期唐氏筛查未见异常;无创产前筛查(Noninvasive Prenatal Test,NIPT)未提示异常。胎儿26+3 周产前超声影像提示大小相当于22+周,长骨短,股骨、肱骨及尺桡骨、胫腓骨长相当于20~21 周(<1%),双顶径(Biparietal Diameter,BPD)60.3 mm(+0.12 SD),头围(Head Circumference,HC)226.5 mm(+0.54 SD),腹围(Abdominal Circumference,AC)188.8 mm(-1.01 SD),股骨长(Femur Length,FL)33.3 mm(-4.17 SD),考虑股骨发育速度明显缓慢,面部轮廓显示额部较前突,鼻梁低平,胸腹部矢状切面显示胸骨内陷。孕妇否认近亲结婚,孕期无放射性物质接触史,否认有家族史,否认孕期药物服用史。孕妇夫妻无异常临床表型,智力均正常。本研究经孕妇及家属的知情同意及本院伦理委员会的审批。
1.2 方法
1.2.1 家系全外显子组测序
抽取脐带血1 mL、胎儿父母外周血2 mL,用EDTA 抗凝剂抗凝。使用商品化核酸提取试剂盒(德国Qiagen)提取EDTA 抗凝血基因组gDNA,测定浓度及纯度A260/A280 在1.8 左右,实验建库采用美国Roche 公司生产的定制基因片段捕获探针(KAPA HyperExome)。实验的标准操作流程包括基因组DNA 打断、磁珠筛选、末端补平、3'端加碱基“A”、PCR 扩增及纯化、目标基因外显子及临近剪切区捕获及杂交、洗涤和洗脱、文库单链环化、高通量测序。测序类型为PE100+10,测序完成后,得到原始测序数据,去除低质量以及被接头污染的读长。将得到的可分析读长用BWA 软件(Burrows Wheeler Aligner)与hg19(GRch37:https://www.ncbi.nlm.nih.gov/assembly/GCF_000001405.13/)进行序列比对。用GATK 软件进行SNV(single nucletide variant)和Indel(insertion and deletion)查询,生成目标区域碱基多态性结果,随后进行多种数据库比对,参考数据库有:NCBI dbSNP v147,dbNSDP v2.9.1,ESP6500 v2 HapMap,1000 human genome dataset 和database of 100 Chinese healthy adults,寻找出可疑的基因变异位点。结合ClinVar(https://www.ncbi.nlm.nih.gov/clinvar/ )、OMIM(https://www.omim.org/)、HGMD(http://www.hgmd.cf.ac.uk/)等疾病数据,参考《美国医学遗传学与基因组学学会遗传变异分类标准与指南2015》[6]规则对变异致病性进行解读。以上检测过程由深圳华大医学检验实验室完成。
1.2.2 Sanger 测序验证
对于在样本发现的CUL7基因、SPINK5基因致病、可能致病变异,在每个变异所在片段的上下游50 bp 附近区域设计引物。进行PCR 扩增,产物进行Sanger 测序,所得结果与NCBI 数据库中CUL7(NM_014780.5)、SPINK5(NM_006846.4)基因标准序列进行比对,从而验证家系外显组测序的结果。
2 结果
2.1 家系全外显子组测序结果
2.1.1CUL7基因的复合杂合变异
CUL7;c.3291_3294delTCAC(p.His1098Cysfs*42)杂合变异,依据ACMG 指南被评判为致病,评判证据为PVS1+PM2+PM3,遗传自母亲。CUL7;c.4717C>T(p.Arg1573*)杂合变异,依据ACMG 指南被评判为致病,评判证据为PVS1+PM2+PM3+PP4,遗传自父亲。见表1。

表1 胎儿家系全外显子组测序结果Table 1 The results of the WES
2.1.2SPINK5基因的复合杂合变异
SPINK5;c.2474_2475delAG(p.Glu825Glyfs*2)杂合变异,依据ACMG 指南被评判为致病,评判证据为PVS1+PM2+PM3,遗传自父亲。SPINK5;c.2870delT(p.Leu957Gln*46)杂合变异,依据ACMG指南被评判为可能致病,评判证据为PVS1+PM2,遗传自母亲。见表1。
2.2 Sɑnger 测序结果
胎儿CUL7;c.3291_3294delTCAC 变异位点为杂合型,母亲为杂合型,父亲为野生型。见图1。胎儿CUL7;c.4717C>T 变异位点为杂合型,母亲为野生型,父亲为杂合型。见图2。胎儿SPINK5;c.2474_2475delAG 变异位点为杂合型,母亲为野生 型,父亲为杂合型。见图3。胎儿SPINK5;c.2870delT 变异位点为杂合型,母亲为杂合型,父亲野生型。见图4。
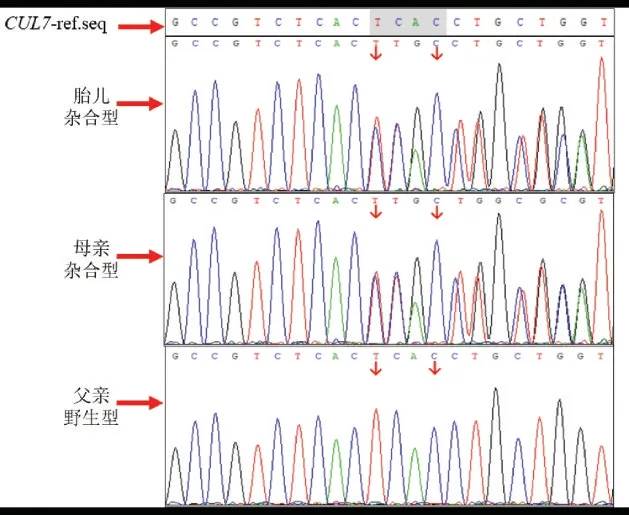
图1 胎儿及其家系CUL7 基因c.3291_3294delTCAC变异位点Sanger 测序图Figure 1 Sanger sequencing showing the c.3291_3294delTCAC of the CUL7 gene in the fetus and parents
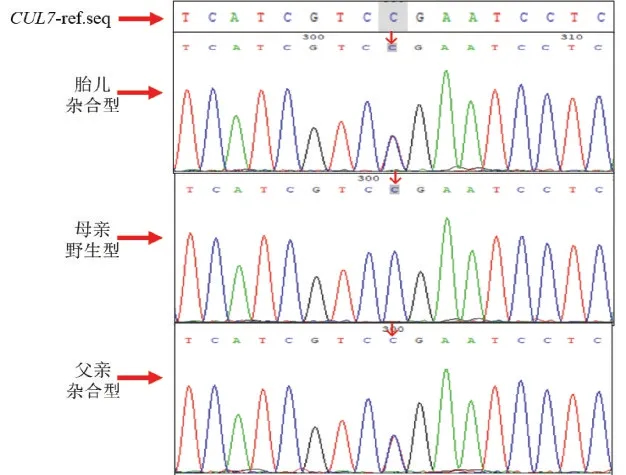
图2 胎儿及其家系CUL7 基因c.4717C>T 变异位点Sanger 测序图Figure 2 Sanger sequencing showing the c.4717C>T of the CUL7 gene in the fetus and parents

图3 胎儿及其家系SPINK5 基因c.2474_2475delAG 变异位点Sanger 测序图Figure 3 Sanger sequencing showing the c.2474_2475delAG of the SPINK5 gene in the fetus and parents

图4 胎儿及其家系SPINK5 基因c.2870delT 变异位点Sanger 测序图Figure 4 Sanger sequencing showing the c.2870delT of the SPINK5 gene in the fetus and parents
3 讨论
据ACMG2020 年发布的胎儿全外显子组测序在产前诊断中的应用显示[7],通过超声鉴定出结构性缺陷的胎儿有30%可能为染色体核型异常,染色体微阵列可以额外检出4%~6%的基因异常;产前全外显子组测序在染色体核型和染色体微阵列正常的情况下额外检出8%~10%的基因异常[8]。宫内发育迟缓是一种胎儿期非特异性的表现,大约占所有活产儿的0.17%,可由多种不同类型的遗传病引起,应在产前进行全外显子组测序提高诊断率[9]。本研究胎儿全外显子组测序结果提示CUL7基因和SPINK5基因分别存在致病/可能致病复合杂合变异,其父母进行变异位点的Sanger测序验证后,证实其父母分别为CUL7基因和SPINK5基因杂合变异携带者。
胎儿CUL7基因c.3291_3294delTCAC 变异和c.4717C>T 变异,其中c.3291_3294delTCAC 为首次报道的致病变异,依据ACMG 指南评判为致病,评判证据项为PVS1+PM2+PM3,SIFT(http://sift.jcvi.org)、MutationTaster(http://www.mutationtaster.org)、PolyPhen(http://genetics.bwh.harvard.edu/pph)等多个蛋白结构/功能和进化保守性软件预测该变异有害,导致蛋白功能缺陷。3M 综合征患者影像学检查表现为骨骼畸形,包括长骨细长、脊柱前凸、椎体高等,表明CUL7参与了骨细胞的生长和增殖。迄今为止,3M 综合征的基因型与表型关系仍未完全清楚,有待进一步的科学研究[10]。
胎儿SPINK5基因c.2474_2475delAG 变异和c.2870delT 变异,其中c.2870delT 变异为首次报道的致病变异,依据ACMG 指南被评判为可能致病,评判证据项为PVS1+PM2,多个蛋白结构/功能和进化保守性软件预测该变异有害,导致蛋白功能缺陷。SPINK5基因位于5q31-32,含有33 个外显子,编码含有1 064 个氨基酸的上皮和粘膜表面的淋巴上皮Kazal 型抑制物(Lymphoepithelial Kazal-Type-Related Inhibitor,LEKTI)蛋白,属于一种多结构域丝氨酸蛋白激酶。有研究表明丝氨酸蛋白酶在脱屑、上皮的抗炎抗菌和皮肤屏障调节中发挥重要作用[11]。NS 患儿在出生时皮肤障碍就严重受损且伴有炎症信号分子显著上升,破坏了皮肤的屏障功能[12]。NS 胎儿出生时即存在弥漫性鳞屑性红皮病样皮损,新生儿期经常出现危及生命的并发症,大多数患儿可以对症治疗,悉心照顾可活至成年[13-15]。
3M 综合征与NS 都是罕见的遗传病,3M 综合征发病率不详;NS 发病率约为1/20 万[16]。胎儿同时发生3M 综合征及NS 的概率极低,本研究为全球首次报道,并且分别增加了一个CUL7基因和一个SPINK5基因新的致病变异,从而扩大了3M 综合征及NS 的基因型谱。NS 患儿的临床表型大多是是出生后才能被发现,产前很难通过超声影像检查发现。引产胎儿外观全身皮肤弥漫性鱼鳞病样红皮,与NS 新生儿皮肤特征相符,但家属拒绝行相关的皮肤检查,所以缺乏NS 疾病的详细临床表型资料。胎儿NS 疾病的发现是因“胎儿宫内发育迟缓、特殊面容”做基因诊断时的额外发现,在遗传咨询过程中,应结合胎儿的宫内发育状况、基因结果、家族史进行客观全面的分析,进行个体化解读,为患者做出最准确的诊断和优生优育咨询。
单基因隐性遗传病的发生,通常是因患儿父母同为遗传病致病基因的携带者。3M 综合征和NS 为不连锁的两种单基因隐性遗传病。按照孟德尔遗传规律,一种隐性遗传病携带的父母生出患儿的概率为1/4,本研究的胎儿父母生出两种疾病患儿的概率为1/16,生出其中一种遗传病患儿的概率为6/16,生出携带者的概率为8/16,生出完全健康的胎儿的概率只有1/16。该孕妇再次妊娠,胎儿为综合征患者的再发风险大大增加,必须进行产前诊断,也可以考虑借助单基因疾病植入前检测(Preimplantation Genetic Testing for Monogenic,PGT-M)辅助生殖技术进行诊断[17]。
猜你喜欢
电子科技大学学报(2022年5期)2022-10-29
中华实用诊断与治疗杂志(2022年1期)2022-08-31
中华实用诊断与治疗杂志(2022年1期)2022-08-31
南京医科大学学报(自然科学版)(2021年8期)2021-10-19
种子(2021年3期)2021-04-12
中国生殖健康(2020年4期)2021-01-18
水产科学(2020年2期)2020-03-20
森林工程(2018年1期)2018-05-14
校园英语·下旬(2017年7期)2017-07-14
科技视界(2016年27期)2017-03-14