
一个两次妊娠外生殖器畸形胎儿家系的临床遗传学分析
2023-02-17 03:01张亚南席惠方芳贾政军庞佳伦邝海燕张巍彭莹
实用医学杂志 2023年1期
张亚南 席惠 方芳 贾政军 庞佳伦 邝海燕 张巍 彭莹
1湖南省妇幼保健院(长沙 410008);2广州嘉检医学检测有限公司(广州 510300)
中国是出生缺陷高发的国家之一,其中先天性外生殖器畸形是一类病因复杂种类繁多的疾病,因其涉及生殖系统的结构和功能,对个人及家庭的影响较其他系统畸形可能更为严重[1]。因此,尽早在产前发现胎儿外生殖器发育畸形及其他相关疾病,明确病因,对准确评估其预后有着重要的临床意义。随着产前超声诊断技术的不断发展,胎儿泌尿系统畸形检出率不断提高,但由于胎儿外生殖器异常病因复杂,很少能在产前得到明确致病原因[2],这也给产前诊断带了新的挑战。
外生殖器的发育因环境、遗传、内分泌等因素使其在胚胎分化时期出现异常分化,表现出不同类型及不同程度的外生殖器畸形[3]。其中以先天性肾上腺皮质增生(congenital adrenal hyperplasia,CAH)、雄激素不敏感综合征和性染色体异常引发的畸形最多见[4-5]。CAH 是一种常染色体隐性遗传病,其中以位于6p21.33 的CYP21A2 基因缺陷引起的21-羟化酶缺乏症(21-hydmxylase deficiency,21-OHD)是最常见的类型[6],CYP21A2 基因编码21 羟化酶,参与肾上腺激素代谢过程[7-8]。21-OHD会引起皮质醇、醛固酮合成不足,女性男性化、男性性早熟,甚至出现肾上腺危象[9-10]。目前21-OHD患者使用的糖皮质激素疗法可能会导致非生理剂量,对健康产生负面后果,患有21-OHD 的女性其生育能力似乎受到了损害[11],尤其是失盐型女患者,发生妊娠糖尿病和剖宫产的风险更高[12]。长效糖皮质激素可能不太有利,尤其是地塞米松[12]。因此,早确诊对预防出生缺陷具有重要意义。
本研究通过联合运用多项产前诊断技术,快速准确寻找一个两次妊娠外生殖器畸形胎儿家系的遗传病因,为该家系的遗传咨询和产前诊断提供了重要参考价值。
1 资料与方法
1.1 一般资料 孕妇,35 岁,G2P0。G1 于2017年因27 周超声提示胎儿外生殖器异常引产,引产胎儿诊断为外生殖器畸形(未做遗传学检测)。本次自然受孕,早孕期无阴道流血,早孕期无感冒等疾病史,无不良因素接触史,本人及家属否认近亲婚配。孕期产检无特殊,未行NT 检测,未行唐氏筛查,NIPT 检测阴性。24 周超声提示胎儿外生殖器异常声像;27 周再次行超声检查,提示胎儿外生殖声像失去正常形态,双侧类似于类阴囊声像不典型。经孕妇知情同意后于28 周行引产前脐血染色体核型分析,尸体解剖和病理检查、基因检测等检查。
1.2 研究方法
1.2.1 脐血染色体核型分析 在超声定位下抽取适量脐带血肝素钠抗凝,按照脐血染色体制备方法进行细胞培养、收获及制片,G 显带,核型分析。常规计数20 个分裂象,分析5 个核型,异常核型时加大计数。根据人类遗传学国际命名体制(ISCN 2020)标准对染色体核型进行命名。
1.2.2 超声学检查 应用GEVoluson E8 彩色多普勒超声诊断仪对胎儿进行系统超声检查,并针对胎儿泌尿生殖器进行详细检查。
1.2.3 尸体解剖 对引产胎儿进行泌尿生殖器器官解剖。
1.2.4 CYP21A2基因检测 采用DNA 提取试剂盒(QIAmp DNA Blood Mini kit,QIAGEN,德国)提取组织/外周血基因组DNA,进行CYP21A2 真基因长片段扩增,排除假基因CYP21A1P 同源序列(同源性高达98%)的干扰,然后将扩增产物进行捕获测序,平均测序深度约为1 000 ×,参考基因组为CYP21A2(NM_000500,hg19)碱基序列,检测点突变和小片段缺失重复。CYP21A2 基因若大片段缺失后会发生基因重组,此时会扩增出一个融合基因的RCCX 模块(RCCX module,RP-C4-CYP21-TNX,包括CYP21A1P 的3 端、所有的C4B 和CYP21A2 的5 端,约30 kb,图1),该模块有3 个酶切位点,此时将CYP21A2 基因长片段扩增产物进行限制性内切酶片段长度多态性分析,检测胎儿组织及胎儿父母CYP21A2 基因30 kb 大片段缺失情况和验证变异来源。若酶切后的电泳图谱只出现3.7/2.5 kb 条带,则没有30 kb 大片段缺失;若检测到主要条带为3.2/2.5 kb,则存在大片段纯合缺失;若酶切图谱中有3.7/2.5 kb 条带和3.2/2.4 kb 条带,则存在大片段杂合缺失。样本外送广州嘉检医学检测有限公司检测。本研究经过医院伦理委员会批准(编号:2020-S085)。该病例的样品采集和遗传学研究获得患者知情同意。
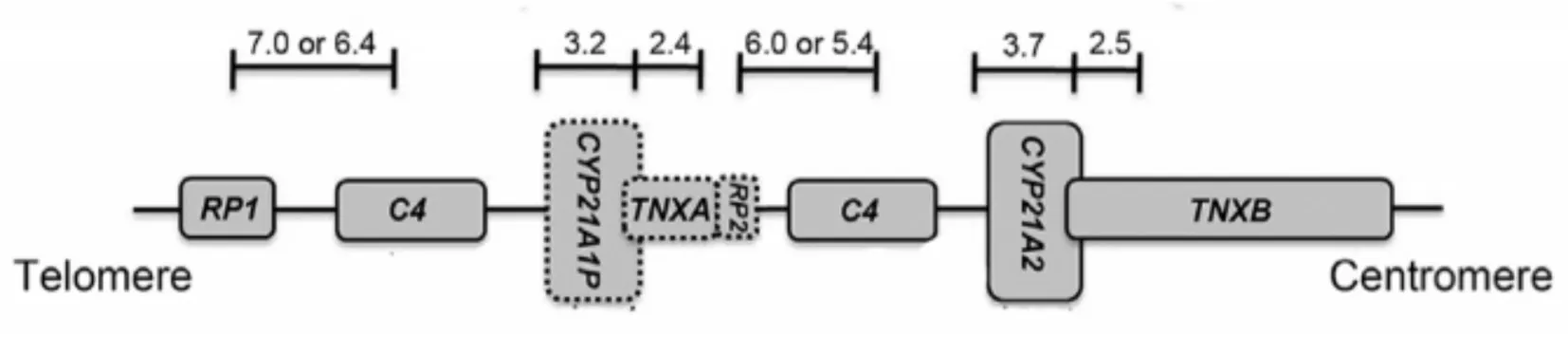
图1 典型的RCCX 模块基因示意图Fig.1 A schematic representation of the typical RCCX module genes
2 结果
2.1 脐血染色体核型分析结果 胎儿脐血染色体核型为46,XX,inv(9)(p11.2q13)(图2),其父母拒绝行外周血染色体核型对照检查。

图2 染色体核型图Fig.2 The karyotype of fetal cord blood cells
2.2 超声学检查 G1(2017年第一次妊娠):27 周超声提示:胎儿外生殖器失去正常声像,未见正常的类阴囊声像,中间可见1.46 cm 长的高回声类阴茎声影,前端朝下,外侧见类大阴唇声像(图3A)。
G2(本研究中即第二次妊娠):24 周超声提示:外生殖声像失去正常形态,双侧类似于类阴囊声像,不典型,中间可见0.9 cm 长的高回声类阴茎声影,末端较扁,朝向肛门方向(图3B)。27 周超声提示:胎儿外生殖声像失去正常形态,双侧类似于类阴囊,声像不典型,末端较扁,朝向肛门方向(图3C)。27 周四维超声提示肾上腺增大(图3D)。
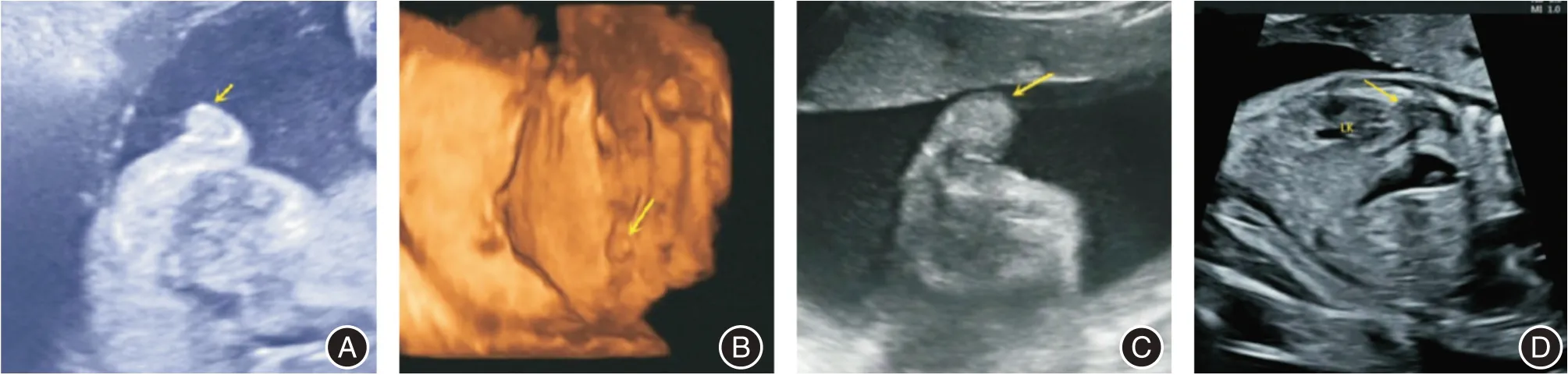
图3 两次妊娠孕晚期胎儿外生殖器声像图Fig.3 Ultrasonography of the genitalia in the third trimester of two pregnancies
2.3 尸体解剖 外生殖器异常(图4A),肾上腺增大(图4B),胎儿其他器官未见其他明显异常。肾上腺病理检查提示胎儿肾上腺皮质微囊性增生(图4C)。
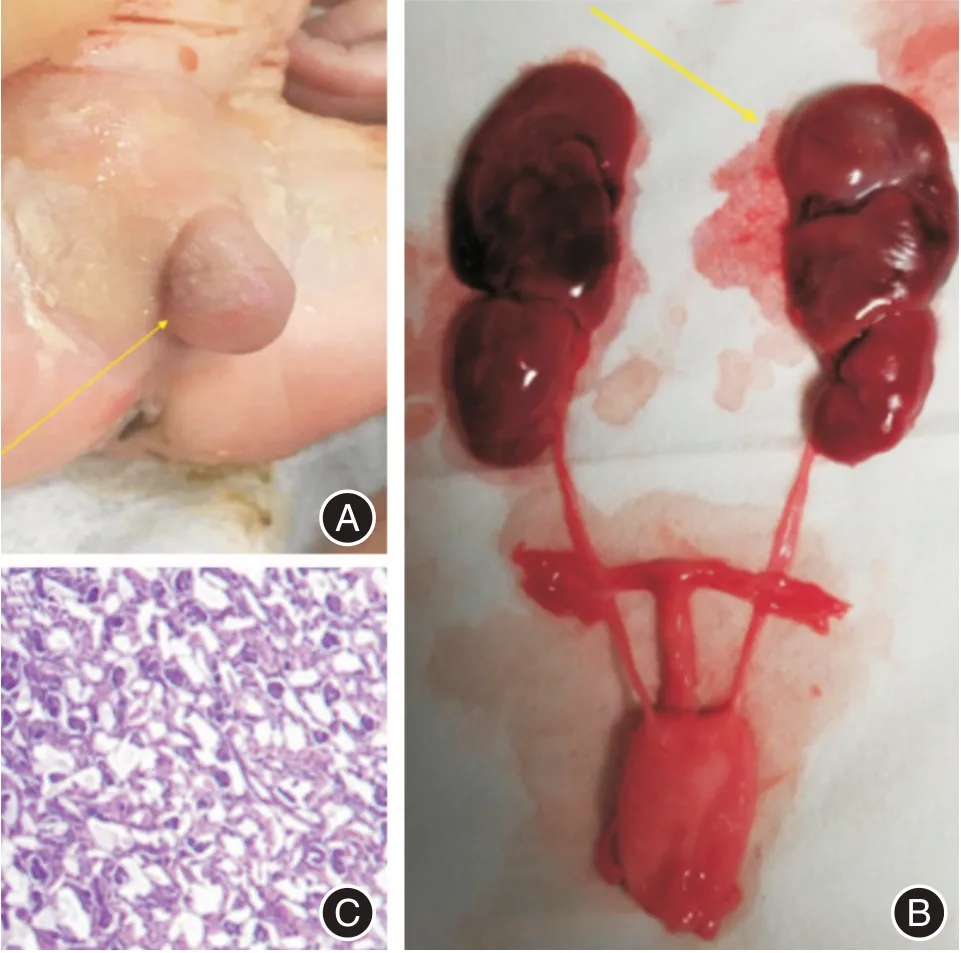
图4 胎儿尸体解剖和病检结果Fig.4 Autopsy and medical examination of the fetus
2.4 CYP21A2 基因检测 根据上述生理和病理检测结果,临床上考虑先天性肾上腺皮质增生,因此对胎儿及其父母进行CYP21A2 基因检测。CYP21A2 基因扩增产物经限制性内切酶片段长度多态性分析,结果提示引产胎儿存在30 kb 大片段纯合缺失,其父母均为携带者(图5)。CYP21A2 基因经长片段PCR 扩增后进行二代测序,检测到11个纯合变异,分别为c.92C > T(p.P31L),c.293-13C > G,c.332_339del8(p.G111Vfs*21),c.518T >A(p.I173N),c.710T > A(p.I237N),c.713T > A(p.V238E),c.719T > A(p.M240K),c.844G > T(p.V282L),c.923dupT(p.L308Ffs*6)、c.955C > T(p.Q319*)和c.1069C > T(p.R357W),这些变异遗传自胎儿的父亲和母亲,提示CYP21A2 基因30 kb纯合缺失形成了CYP21A1P/CYP21A2 嵌合基因,从而导致CYP21A2 基因失活。CYP21A1P/CYP21A2融合基因具体9 种分型依据[13],本例胎儿携带大片段纯合缺失,存在p.P31L、c.293-13C > G、c.332_339del8、p.I173N、p.Q319*和p.R357W 变异,因此CYP21A1P/CYP21A2 融合基因类型是CH-8 型。
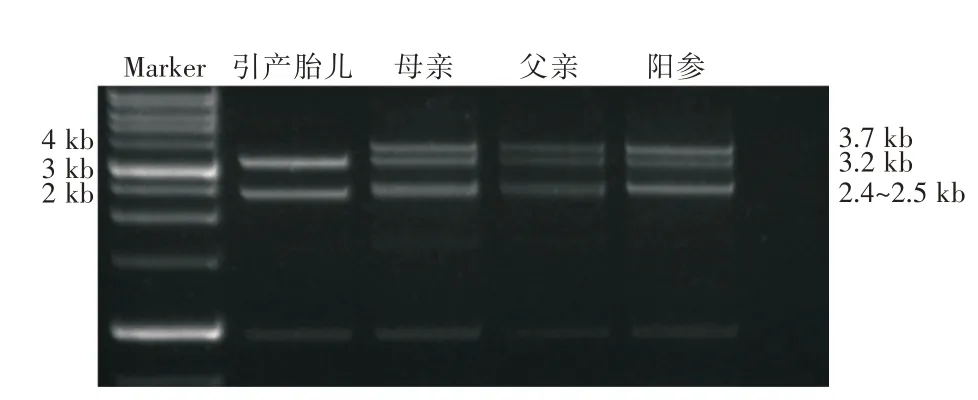
图5 CYP21A2 基因长片段扩增产物进行限制性内切酶片段长度多态性检测Fig.5 Restriction endonuclease fragment length polymorphism was detected by long fragment amplification products of CYP21A2 gene
3 讨论
本研究中该家系两次妊娠,均在孕中期超声检查发现胎儿外生殖器异常,最终均选择了引产。首先通过脐血染色体核型分析,确定胎儿染色体为46,XX,inv(9)(p11q13);同时超声检查提示胎儿外生殖器畸形和肾上腺增大,尸检结果与超声检查一致。鉴于以上结果显示生殖器和肾上腺异常,临床上考虑引产胎儿患有21-羟化酶缺乏症,于是获得家属知情同意后进行基因测序,最终诊断胎儿为CAH 患者。21-羟化酶缺乏会导致皮质醇和醛固酮合成异常,促肾上腺皮质激素代偿性分泌增加,引起肾上腺皮质增生及雄激素合成增多。21-OHD 主要因CYP21A2 基因大片段缺失和基因转换引起[14],目前已经报道与21-OHD 有关的突变有1 300 多种,不同的基因突变导致不同程度21-羟化酶活性下降,引起21-OHD 不同临床表型[15-17],也是引起女性胎儿外生殖器男性化畸形最常见的原因。46,XX 胎儿在胚胎时期由于肾上腺源性雄激素过多,其外生殖器在胚胎6~8 周出现男性化改变,如阴蒂增大、阴唇融合、尿生殖窦形成[8,18]。
随着遗传学技术的快速发展,全外显子测序因检测深度大,范围广等优势已广泛运用在临床罕见病、疑难病中。但对于本研究所涉及的21-OHD 而言,由于其致病基因CYP21A2 存在一个与其基因序列高度同源的假基因(CYP21A1P),其外显子和内含子的同源性分别高达98%和95%,且主要致病突变是真假基因间的高频率的基因转换和重排导致[7,19],故全外显子测序对于该病的检测并不适用,目前常用的检测方法是Sanger 测序联合多重连接探针扩增(MLPA)[6,20]、设计序列特异性引物进行CYP21A2 基因扩增联合下一代测序(NGS)进行测序[21]等方法将基因点突变、大片段缺失和重复同时检出,避免漏诊。
本文在对引产胎儿及其父母进行CYP21A2 基因突变检测和分析中,采用限制性内切酶片段长度多态性分析,发现胎儿存在一个30 kb 大片段纯合缺失变异,其父母均为非近亲的携带者;同时设计引物将CYP21A2 真基因长片段扩增,检测到胎儿存在11 个纯合变异,分别为c.92C>T(p.P31L),c.293-13C > G,c.332_339del8(p.G111Vfs*21),c.518T>A(p.I173N),c.710T>A(p.I237N),c.713T >A(p.V238E),c.719T > A(p.M240K),c.844G > T(p.V282L),c.923dupT(p.L308Ffs*6)、c.955C > T(p.Q319*)和c.1069C>T(p.R357W),这些变异遗传自胎儿的父亲和母亲。因此,发现该胎儿CYP21A2基因存在11 个纯合变异是由30 kb 纯合缺失相关的同源基因重组引起,且这些突变均能造成不同程度21-羟化酶活性下降,从而导致CYP21A2 基因失活。研究表明,CYP21A2 基因大片段缺失的突变类型占20%[22],缺失范围为从CYP21A1P 假基因3'端到CYP21A2 基因5'端,大部分突变遗传自父母,可导致21-羟化酶活性完全丧失,是21-OHD最常见的致病基因突变之一。本研究引产胎儿还携带致病性最强的变异p.(V282L)[20]。CYP21A2不同的基因突变造成不同程度21-羟化酶活性下降,引起不同的临床表型[23],其中典型2l-OHD 女童的男性化程度与CYP21A2 基因变异导致的酶活性丧失程度呈正相关[24]。本研究胎儿在孕期就出现明显的外生殖器异常,是因为CYP21A2 基因30 kb 大片段纯合缺失造成真假基因融合,产生融合基因CH-8 型,严重损害蛋白质的结构和功能,造成21-羟化酶的酶活性极度缺乏或完全丧失[14],导致胎儿皮质激素合成不足,增大促肾上腺素分泌,大量皮质醇前体产物堆积并向雄性激素转化,最后表现出肾上腺增大和外生殖器异常,胎儿出生后还可能会出现肾上腺危象、休克和后遗症。
本研究通过对一个两次妊娠外生殖器畸形胎儿家系先后联合运用脐血染色体核型分析、产前超声检查以及尸体解剖验证,层层递进,环环相扣,有针对性的选择了CYP21A2 基因检测方案。最终发现胎儿携带的CYP21A2 基因突变是21-羟化酶缺乏症的致病原因,从而明确该家系两次妊娠外生殖器畸形胎儿的遗传病因,为其遗传咨询和产前诊断提供了重要依据,对降低出生缺陷、优生优育有重要指导意义。本研究也存在不足之处,由于基因检测只针对CYP21A2 基因,胎儿期表型观察有限,不排除本报道高龄产妇携带其他基因致病突变,建议夫妻生育前进行携带者筛查。
猜你喜欢
中华实用诊断与治疗杂志(2022年2期)2022-09-02
宁夏医学杂志(2020年3期)2021-01-21
热带作物学报(2020年9期)2020-10-29
中国现代医生(2020年12期)2020-07-04
甘肃农业科技(2019年10期)2019-09-10
保健与生活(2018年20期)2018-01-27
西南军医(2016年5期)2016-01-23
江苏农业科学(2015年10期)2015-12-23
哈尔滨医药(2015年2期)2015-12-01
中国当代医药(2015年9期)2015-03-01
