
基于极大团扩展的蛋白质复合物识别算法
2010-05-31 06:09李敏王建新刘彬彬陈建二
中南大学学报(自然科学版) 2010年2期
李敏,王建新,刘彬彬,陈建二
(中南大学 信息科学与工程学院,湖南 长沙,410083)
随着人类基因组计划和许多物种全基因组测序的完成,生命科学研究的重点逐渐从基因组学转移到蛋白质组学[1-2]。蛋白质是一切生命活动的物质基础[3]。但细胞中每个蛋白质并不是孤立存在的,而是通过与其他蛋白质相互作用形成大的蛋白质复合物来行使其功能的[4]。1个生物体内所有蛋白质相互作用被称为蛋白质相互作用网络(Protein interaction network)。从大规模相互作用网络中识别蛋白质复合物对预测蛋白质功能、解释特定的生物进程具有重要作用。目前,出现了一些较有效的蛋白质复合物识别算法,如:基于图划分的 RNSC算法[5],基于密度的局部搜索算法MCODE[6],基于边介数的G-N算法[7]和在其基础上改进的 MoNet算法[8]等。虽然这些算法各有各的优点,但都存在1个共同的缺点,就是不能识别交叠蛋白质复合物(Overlapping protein complexes)。而在实际的蛋白质相互作用网络中,每个蛋白质可能属于多个蛋白质复合物。例如,在CYGD数据库[9]中由2 750个蛋白质组成的复合物中蛋白质的数量之和为8 932。所以,研究交叠蛋白质复合物的识别算法具有实际意义。Palla等[10]研究了一种基于团渗透的算法(Clique percolation method,CPM),用来分析包括蛋白质相互作用网络在内的复杂网络的交叠和嵌套结构。2006年,该团队开发了基于 CPM 算法的复杂网络分析工具 CFinder[11],这是目前识别交叠蛋白质复合物最有效的工具。但是,算法CPM的识别结果与参数k的取值密切相关,识别的蛋白质复合物数量有限,特别是k取值比较大时能够识别的蛋白质复合物数量就更少,而当k取值较小时,CPM算法通常会产生规模比较庞大的复合物,而这样的复合物往往包含了规模远大于k的团结构和比较稀疏的k-团链。在实际应用中,更希望将这种复合物分割成多个比较稠密的子图。为解决这一问题,本文作者提出了1种基于极大团扩展的蛋白质复合物识别算法(IPC-MCE),将极大团看作蛋白质复合物的核,通过考查核的邻居顶点与核内顶点的作用概率决定邻居顶点是否属于该复合物。将算法应用于酵母蛋白质相互作用网络。实验结果表明:算法 IPC-MCE能够识别较多的具有生物意义的蛋白质复合物,且对参数不敏感。
1 算法IPC-MCE
蛋白质相互作用网络可以被表示成为1个无向简单图G=(V,E),V和E分别为图G的顶点集和边集,图G中的每个顶点表示1个蛋白质,每条边表示1对蛋白质之间的相互作用。Palla等[10]提出的CPM算法将蛋白质复合物看作图中相互连通的若干k-团集合。k-团是指包含k个顶点的全连通图。若2个k-团有k-1个公共顶点,则称这2个k-团是邻接的。一系列邻接的k-团组成1个k-团链。若2个k-团出现在1个k-团链中,则称这2个k-团是连通的。CPM算法通过迭代递归得到图中所有的k-团,然后,建立这些团的交叠矩阵,通过分析交叠矩阵得到各个连通的k-团集合。显然,k的取值越大,CPM算法得到的每个k-团集合中所包含的k-团个数就越少;相反,当k取值很小时,则很容易形成巨大的k团集合。鉴于此,本文提出了1种新的基于极大团扩展的蛋白质复合物识别算法IPC-MCE。
与CPM算法不同,IPC-MCE算法将蛋白质复合物看作1个以极大团为核、其他顶点与核紧密相连的稠密子图,并提出用作用概率来量化其他顶点与核连接的紧密程度,定义如下。
定义1 作用概率IPvk:顶点v与极大团k的作用概率|表示极大团的顶点数,|Evk|表示顶点v与极大团k内顶点之间存在的边数。
算法IPC-MCE的描述如图1所示。

图1 IPC-MCE算法描述Fig.1 Description of IPC-MCE algorithm
算法 IPC-MCE的输入是蛋白质相互作用信息(PPI)。算法首先根据这些蛋白质相互作用信息建立网络图G。考虑到蛋白质相互作用网络中存在大量的度为1的顶点,而这些度为1的顶点只能构成规模为2的极大团,对其进行扩展只能生成以其邻居顶点为中心的HUB结构子图。所以,IPC-MCE算法在枚举图中所有极大团之前先对这些度为 1的顶点进行预处理,在原图G中删除这些度为1的顶点及其连接的边,并将这些1度顶点及对应的邻居顶点组成的HUB结构子图输出。图2给出了几个HUB结构子图的实例。
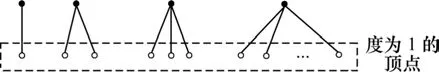
图2 蛋白质相互作用网络中的HUB结构子图实例Fig.2 Examples for subgraphs of protein interaction networks which have HUB structures
预处理后,IPC-MCE算法枚举残图中所有的极大团。采用Tsukiyama等[12]提出的基于深度优先的极大团枚举算法,首先选择图中度最大的顶点,寻找其对应的所有极大团,然后选择度次大的顶点,寻找其对应的所有极大团,依此类推。该极大团枚举算法可以通过横向和纵向剪枝来提高算法效率,其时间复杂度为)(μnmΟ,其中:n为图的顶点个数;m为图边数;μ为极大团个数。
然后,根据定义1的作用概率IPvk,IPC-MCE算法对枚举得到的所有极大团进行局部扩展。考虑每个极大团的一阶邻居顶点(即那些与极大团内的顶点有直接相互作用且不在极大团内的顶点),判断这些邻居顶点是否满足IPvk≥t,将满足条件的邻居顶点扩展进来,得到扩展后的稠密子图。对扩展得到的所有稠密子图,过滤掉其中重复的稠密子图,得到最终的稠密子图集合DS并输出,整个IPC-MCE算法运行结束。
算法中步骤(1)的时间复杂度为 O(m);步骤(2)的时间复杂度为O(n);步骤(3)枚举极大团的时间复杂度为)(μnmΟ;步骤(4)极大团扩展的时间复杂度为Ο(nsμ);步骤(5)的时间复杂度为Ο(μ2d2)。其中:μ为规模大于等于3的极大团个数;s为最大的极大团规模;d为最大的稠密子图规模。
2 实验结果及分析
用C++语言实现IPC-MCE算法,从http://angel.elte.hu/clustering/下载基于CPM算法开发的交叠蛋白质复合物识别工具CFinder。这2个算法的测试是在1台安装了Windows XP Professional操作系统的工作站(Intel Pentium 1.73 GHz,内存为512 MB)上进行。
2.1 实验数据
实验以酵母蛋白质相互作用网络作为研究对象,因为酵母是所有物种中蛋白质相互作用数据最为完备的。实验所用的蛋白质相互作用数据和用于评估的标准蛋白质复合物数据集来源于MIPS数据库[13]。在数据预处理阶段,去除了蛋白质相互作用网络中自相互作用和冗余的相互作用,最终的相互作用网络包括4 546个酵母蛋白质和12 319对相互作用。将包含2个及以上蛋白质的216个复合物作为标准复合物数据集。最小的复合物包括2个蛋白质,最大的复合物包括81个蛋白质,平均每个复合物包括6.31个蛋白质。
2.2 性能评价
目前,对蛋白质复合物识别算法的性能主要是从算法标识已知复合物的能力,以及算法的敏感度和特异性这几个方面进行评价[6,11]。下面从这几个方面对算法IPC-MCE和CFinder的性能进行对比分析。
2.2.1 已知蛋白质复合物被标识的数量
算法识别的蛋白质复合物(Predicted complexes,Pc)与已知蛋白质复合物(Known complexes, Kc)的匹配程度OS(Pc, Kc)的计算公式[5,9-10]为:
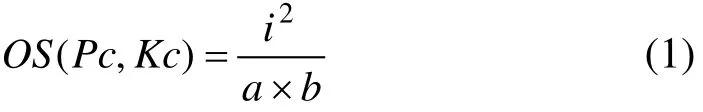
其中:a和b分别表示Pc和Kc所包含的蛋白质个数,i表示Pc和Kc中公共蛋白质个数。
若 2个蛋白质复合物的匹配程度 OS(Pc,Kc)超过给定的阈值,则称这2个复合物匹配。对于标准复合物数据集中的已知复合物,若识别出的复合物与之匹配程度OS(Pc, Kc)超过给定阈值,则称该已知复合物被标识,若OS(Pc, Kc)=1,则称该已知复合物被完全标识。算法标识的已知复合物数量越多,说明算法识别复合物的能力越强。
图 3描述了不同匹配阈值下已知复合物被 IPCMCE算法和CFinder标识的数量。图3中CFinder (k=3)和CFinder (k=4)分别表示CFinder在参数k为3和4时标识的已知复合物数量;IPC-MCE(DS, t=*)表示算法IPC-MCE在参数t取不同值时输出的稠密子图标识的已知复合物数量。IPC-MCE(HUB&DS, t=0.8)表示算法IPC-MCE在参数t=0.8时输出的HUB结构子图和稠密子图一起标识的已知复合物数量。
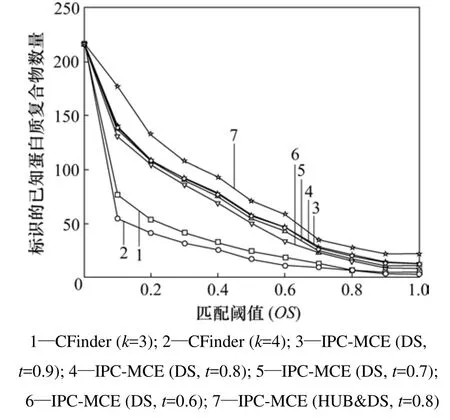
图3 不同匹配阈值下已知复合物被标识的数量Fig.3 Number of matched known complexes with respect to different overlapping score thresholds
从图3可以看出:在不同匹配程度阈值下,算法IPC-MCE在t=0.6,0.7,0.8和0.9时产生的稠密子图结果数据集标识的已知复合物数量相差不多,说明算法的结果数据集对输入的参数不敏感。
CFinder在k=3时标识的已知复合物数量最大,随着k增大,CFinder在不同匹配程度阈值下能够标识的已知复合物数量逐渐下降。这是因为随着k增大,基本的拓扑单元k-团随之减少,能够识别的蛋白质复合物数量也随之减少。ZHANG等[14-15]利用 CFinder分析蛋白质相互作用网络也给出了相同的结论。在实验中,CFinder在k=3,4,5,6和7时识别出来的蛋白质复合物数量分别为178,61,18,9和8。从图3可以看出:算法 IPC-MCE标识的已知复合物数量明显高于Cfinder标识的数量。此外,算法IPC-MCE预处理产生的 HUB结构子图对标识已知复合物也有贡献。几乎所有 CFinder能够识别的蛋白质复合物IPC-MCE都能够准确地识别出来。图4给出了几个具体的实例。
图4(a)中,算法IPC-MCE 在t=0.8时完全标识的1个规模为10的蛋白质复合物,CFinder在k=3时识别的与该已知复合物最佳匹配程度OS(Pc,Kc) =0.556。对图4(a)中CFinder识别的复合物,算法IPC-MCE将其分割为几个相互交叠的复合物。
图4(b)中算法IPC-MCE 在t=0.8时识别的1个规模为 14的蛋白质复合物与已知复合物的匹配程度OS(Pc, Kc)=0.875,CFinder在k=3时识别的所有复合物与该已知复合物匹配程度OS(Pc, Kc)<0.1,在k=4时识别的复合物与该已知复合物最佳匹配程度OS(Pc,Kc) =0.583。这说明CFinder识别的复合物的稠密程度与k的取值密切相关,随着k增大,CFinder更容易识别比较稠密的复合物。
图4(c)中算法IPC-MCE 在t=0.5时识别的1个规模为 7的蛋白质复合物与已知复合物的匹配程度OS(Pc, Kc)= 0.735,CFinder在k=3时识别的与该已知复合物最佳匹配程度 OS(Pc, Kc)=0.643。这说明:对于比较稀疏的复合物,算法IPC-MCE比CFinder更有效地识别。
为进一步测试算法IPC-MCE和CFinder识别复合物的能力,将这2个算法识别的复合物与Gavin等[16-18]使用串联亲和纯化和大规模质谱分析等系统分析方法得到的复合物进行对比分析。Gavin,Ho和 Krogan所得的数据集分别包含232,552和91个蛋白质复合物。在判断2个复合物是否匹配时,通常将匹配程度OS(Pc, Kc)的阈值设置为0.2[6]。图5描述了MIPS等数据集中被算法IPC-MCE和CFinder识别的复合物所匹配(OS(Pc, Kc)≥0.2)的已知复合物数量。从图5可以看出:算法IPC-MCE不仅能够标识出大量的MIPS数据库中人工注释的蛋白质复合物,而且也能够标识出比较多的大规模系统分析得到的蛋白质复合物,并且算法IPC-MCE在t=0.8时识别的复合物所匹配的4个数据集中复合物的数量远高于CFinder在k=3时识别的复合物所匹配的数量。这说明:算法 IPC-MCE比CFinder 具有更强的蛋白质复合物识别能力。
2.2.2 算法的特异性和敏感度
算法的特异性P和敏感度R是用来评估蛋白质复合物识别算法的另外 2个重要指标[6]。特异性是指算法识别的蛋白质复合物中识别正确的部分所占比例,如式(2)所示;敏感度是指已知蛋白质复合物中被算法识别出来的部分所占比例,如式(3)所示。式中:T (True positive)表示算法识别的复合物中与已知复合物匹配程度值OS(Pc, Kc)≥0.2的个数;F (False positive)等于识别的复合物总数减去T;N (False negative)表示已知复合物中没有识别的复合物与之匹配程度OS(Pc, Kc)≥0.2的个数。

图4 算法IPC-MCE和CFinder在参数不同取值下识别的复合物以及标识出来的已知复合物Fig.4 Examples for protein complexes identified by IPC-MCE and CFiner and corresponding matched known complexes
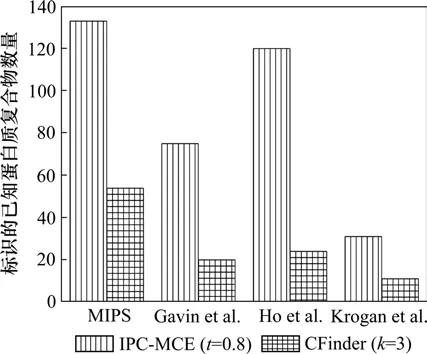
图5 OS(Pc, Kc)≥0.2时已知复合物被标识的数量Fig.5 Number of matched known complexes with respect to OS(Pc, Kc)≥0.2
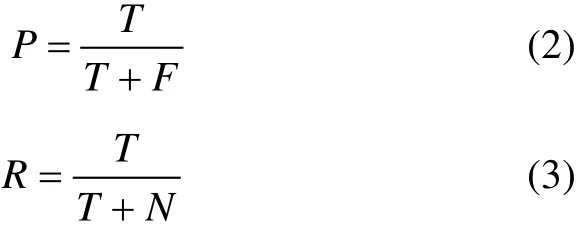
Li等[19]综合考虑敏感度和特异性2个方面,提出综合评价指标f,其计算公式[19]如下:

算法IPC-MCE和CFinder的特异性P、敏感度R和综合评价f的比较结果如表1所示。
从表1可以看出:算法IPC-MCE在t=0.6,0.7,0.8和0.9时识别的复合物的敏感度在0.8以上,说明识别出来的复合物与已知复合物匹配阈值大于等于0.2的数量(T)远高于已知复合物在该阈值下没有被标识的数量(N),前者是后者的4倍多。T越大,N越小,敏感度就越高,说明算法识别出来的复合物可靠性越高。CFinder的敏感度最高只有0.213 6。算法IPC-MCE在t=0.6,0.7,0.8和0.9时识别的复合物的特异性大于0.23,而CFinder的特异性都大于0.24,明显高于算法IPC-MCE的特异性。虽然CFinder的特异性高于IPC-MCE的特异性,但CFinder识别的复合物中与已知复合物匹配的数量(T)却远小于 IPC-MCE匹配的数量。此外,由于目前试验测定的已知蛋白质复合物数据不完整,算法识别的蛋白质复合物中可能存在一定比例的复合物还未被试验测定但是真实存在。从2个算法的综合评价指标f可以看出:算法IPC-MCE的f值要远高于Cfinder的f值,说明算法IPC-MCE在识别蛋白质复合物方面比CFinder的性能好。
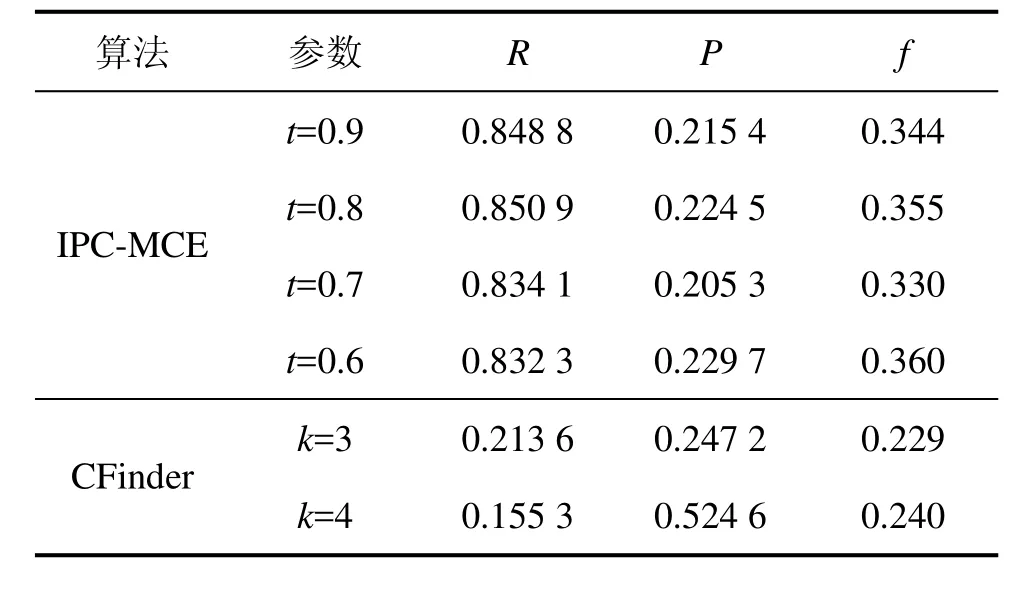
表1 算法 IPC-MCE与CFinder的敏感度R、特异性P和综合评价f的比较结果Table 1 Comparison of IPC-MCE and CFinder under Sensitivity, Specificity, and f-measure
2.3 功能富集分析
很多研究者根据超几何聚集分布的 P-value来注释识别复合物的主要功能,其计算式[8]如下:

其中:n1和n2分别表示识别的复合物和功能注释复合物的规模;i表示他们交集的规模;N表示网络的规模。
P-value 体现了识别复合物中蛋白质功能富集的概率。一般,将 P-value的最小值对应的功能作为该复合物的主要功能。但很多蛋白质复合物的注释功能至少对应于2种功能类。这里,采取文献[5, 14]中所用的Pol计算方法,通过最小化识别复合物与功能注释复合物的随机交叠概率来获得识别复合物的最佳匹配功能注释复合物:

其中:n1和n2分别表示识别的复合物和功能注释复合物的规模;i表示他们交集的规模;N表示网络的规模。
将IPC-MCE算法在t=0.8时识别的规模大于5的442个蛋白质复合物与功能注释复合物进行比较,计算Pol,发现超过98.4%的Pol小于0.01,其中约86.2%的Pol小于0.001,超过72.6%的Pol更小,小于1×10-10。CFinder在k=3时识别的规模大于5的蛋白质复合物只有 29个,Pol小于 0.01的占 93.1%,而 Pol小于1×10-10的仅有16个,占55.2%。上述分析说明算法IPC-MCE识别的蛋白质复合物不仅从统计上证明是具有生物意义的,并且较 CFidner具有更强的生物意义。
3 结论
(1) 与交叠蛋白质复合物识别工具 CFinder的识别结果比较,提出的 IPC-MCE算法在相同条件下能够更精确地标识已知蛋白质复合物。在最优参数设置下,IPC-MCE算法标识的已知蛋白质复合物数量是CFinder标识的已知蛋白质复合物数量的2倍多,说明算法IPC-MCE具有更好的识别性能。
(2) 基于Pol的统计分析表明:算法IPC-MCE能够识别出蛋白质网络中具有生物意义的蛋白质复合物。统计分析规模大于5的蛋白质复合物的Pol发现,98.4%的Pol小于0.01。
(3) 算法IPC-MCE不需要其他任何辅助信息,简单有效,且对输入参数不敏感,能够为蛋白质复合物识别和蛋白质功能预测提供有益的参考。
[1] Graves P R, Haystead T A. Molecular biologist’s guide to proteomics[J]. Microbiol Mol Biol Rev, 2002, 66(1): 39-63.
[2] 向亚莉, 易红, 李萃, 等. 鼻咽癌细胞系5-8F和6-10B的差异蛋白质组学研究[J]. 中南大学学报: 医学版, 2007, 32(6):978-984.XIANG Ya-li, YI Hong, LI Cui, et al. Differential proteomic analysis of naspharyngeal carcinoma cell lines 5-8F and 6-10B[J]. Journal of Central South University: Medicine Science,2007, 32(6): 978-984.
[3] CHENG Yun-hui, WANG Zhang, XU Shi-ying. Antioxidant properties of wheat germ protein hydrolysates evaluated in vitro[J]. Journal of Central South University of Technology,2006, 13(2): 160-165.
[4] Gavin A C, Superti-Furga G. Protein complexes and proteome organization from yeast to man[J]. Curr Opin Chem Biol, 2003,7(1): 21-27.
[5] King A D, Pržulj N, Jurisica I. Protein complex prediction via cost-based clustering[J]. Bioinformatics, 2004, 20(17):3013-3020.
[6] Bader G D, Hogue C W. An automated method for finding molecular complexes in large protein interaction networks[J].BMC Bioinformatics, 2003, 4: 2.
[7] Girvan M, Newman M. Community structure in social and biological networks[J]. PNAS, 2002, 99: 7821-7826.
[8] LUO Feng, YANG Yue-feng, CHEN Chin-fu, et al. Modular organization of protein interaction networks[J]. Bioinformatics,2007, 23(2): 207-214.
[9] Güldener U, Münsterkötter M, Kastenmüller G, et al. CYGD: the comprehensive yeast genome database[J]. Nucleic Acids Res,2005, 33: D364-D368.
[10] Palla G, Dernyi I, Farkas I, et al. Uncovering the overlapping community structure of complex networks in nature and society[J]. Nature, 2005, 435: 814-818.
[11] Adamcsek B, Palla G, Farkas I, et al. CFinder: locating cliques and overlapping modules in biological networks[J].Bioinformatics, 2006, 22(8): 1021-1023.
[12] Tsukiyama S, Ide M, Ariyoshi H, et al. A new algorithm for generating all the maximal independent sets[J]. SIAM Journal on Computing, 1977, 6(3): 505-517.
[13] Mewes H W. MIPS: analysis and annotation of proteins from whole genome in 2005[J]. Nucleic Acid Research, 2006, 34:169-172.
[14] ZHANG Shi-hua, NING Xue-mei, ZHANG Xing-sun.Identification of functional modules in a PPI network by clique percolation clustering[J]. Computational Biology and Chemistry.2006, 30(6): 445-451.
[15] Jonsson P, Cavanna T, Zicha D, et al. Cluster analysis of networks generated through homology: Automatic identification of important protein communities involved in cancer metastasis[J]. BMC Bioinformatics, 2006, 7: 2.
[16] Gavin A C, Bosche M, Krause R, et al. Functional organization of the yeast proteome by systematic analysis of protein complexes[J]. Nature, 2002, 415: 141-147.
[17] Ho Y, Gruhler A, Heilbut A, et al. Systematic identification of protein complexes in saccharomyces cerevisiae by mass spectrometry[J]. Nature, 2002, 415: 180-183.
[18] Krogan N J, Peng N J, Cagney G, et al. High-definition macromolecular composition of yeast RNA- processing complexes[J]. Molecular Cell, 2004, 13: 225-239.
[19] Li X L, Tan S H, Foo C S, et al. Interaction graph mining for protein complexes using local clique merging[J]. Genome Informatics, 2005, 16(2): 260-269.
猜你喜欢
中等数学(2021年9期)2021-11-22
中等数学(2021年8期)2021-11-22
黑龙江科学(2021年14期)2021-08-06
同济大学学报(自然科学版)(2019年2期)2019-04-02
计算机与数字工程(2019年3期)2019-03-26
中成药(2018年7期)2018-08-04
中成药(2018年3期)2018-05-07
中成药(2017年5期)2017-06-13
科技视界(2013年23期)2013-08-22
植物营养与肥料学报(2012年5期)2012-10-26
