
氧化铝载体和氧化钡助剂对钌基氨合成催化剂结构和性能的影响
2010-11-30 10:49杨晓龙夏春谷唐立平熊绪茂慕新元
物理化学学报 2010年12期
杨晓龙 夏春谷 唐立平 熊绪茂 慕新元 胡 斌,*
(1中国科学院兰州化学物理研究所,兰州 730000; 2中国科学院研究生院,北京 100049)
氧化铝载体和氧化钡助剂对钌基氨合成催化剂结构和性能的影响
杨晓龙1,2夏春谷1唐立平1,2熊绪茂1慕新元1胡 斌1,*
(1中国科学院兰州化学物理研究所,兰州 730000;2中国科学院研究生院,北京 100049)
采用不同来源γ-Al2O3(市售Al2O3-1,合成Al2O3-2)作为钌基氨合成催化剂载体,利用浸渍法制备了一系列添加不同BaO助剂含量的Ba-Ru/Al2O3催化剂.通过X射线衍射(XRD)、N2-低温物理吸附、X射线荧光光谱(XRF)、透射电镜(TEM)、H2程序升温还原(H2-TPR)、NH3程序升温脱附(NH3-TPD)和X射线光电子能谱(XPS)等方法研究了不同来源的Al2O3以及BaO助剂含量对负载型钌基催化剂的物相结构、织构性质、微观形貌、表面性质和催化剂的氨合成活性等方面的影响.结果表明,载体的物理化学性质对制备的钌基氨合成催化剂的结构以及活性有较大影响.BaO助剂对催化剂的影响主要表现在两个方面:添加量不同导致BaO与γ-Al2O3的作用力不同,从而进一步影响催化体系的比表面积和孔结构性质;BaO助剂会对体系的Ru物种还原性质以及催化剂表面酸碱性质进行调节,适量BaO的加入能够极大提高反应活性,而这种最佳量与载体性质密切相关.
氨合成;γ-Al2O3;载体;Ru;还原;酸性
钌基氨合成催化剂被誉为新一代氨合成催化剂,其研究与开发成为当今国内外学者关注的热点之一[1-10].与铁基催化剂相比,钌基催化剂的主要优点是低温低压活性高,对水、CO和CO2不敏感,受NH3的抑制作用不明显,可在较高的氨浓度下操作,因此是理想的低温低压氨合成催化剂[2].英国BP公司于1979年开发了石墨化活性炭负载钌基氨合成催化剂,之后,BP公司和美国Kellogg公司合作,于1990年成功开发出全球第一套以石墨化活性炭负载钌基氨合成催化剂为基础的KAAP(Kellogg Advanced Ammonia Process)工艺流程.投产后的运行情况表明钌基催化剂的寿命与活性炭载体的流失密切相关,在氨合成反应条件下,Ru能催化活性炭甲烷化反应.因此,活性炭载体的稳定性及其在工业使用过程中的流失是一个重要的问题.此外,活性炭载体的石墨化过程条件苛刻、生产成本较高、操作与控制比较复杂[2,4],这些因素极大地阻碍了钌基催化剂的工业化应用进程.
针对这些问题,寻找优良载体成为新一代钌基氨合成催化剂研究开发的关键,载体应在氨合成条件下具有较高的稳定性,一定的机械强度,合适的比表面积和孔结构.目前,一些难还原的金属氧化物,如Al2O3[4,911]、MgO[6,8,12]、MgAl2O4[5]等已被广泛研究.通过对金属氧化物载体负载钌基氨合成催化剂的系统研究,Aika等[3]认为载体的电负性越小(即碱性越强),催化剂的活性越高.Al2O3作为一种最常用的载体,由于表面存在较多的Lewis酸性中心,因此氨合成活性相对较低,被普遍认为不适于做钌基催化剂载体.张新波等[4]认为,Al2O3具有比表面积较高、化学性质稳定和机械强度较好等优点,可以通过修饰或改变其表面性质(如降低其表面酸性,提高表面导电性)等方法,提高Al2O3负载钌基催化剂的氨合成活性.虽然,Al2O3具有成为工业化钌基氨合成催化剂载体的潜力,然而,目前国内对Al2O3负载钌基催化剂的详细研究报道还不多[4,13-14].我们从两个方面对Al2O3负载钌基催化剂开展系统研究: γ-Al2O3载体和BaO助剂对钌基催化剂结构和氨合成活性的影响.探索不同来源Al2O3作为钌基氨合成催化剂载体的异同点,BaO助剂的促进作用如何受载体的影响等,探讨Al2O3载体和BaO助剂影响催化剂性质和氨合成活性的本质原因.
1 实验
1.1 载体的合成
工业用氧化铝(γ型,GA-385,江苏省三剂实业有限公司)在500°C马弗炉中焙烧6 h,研磨备用,记为Al2O3-1.将一定量的Al(NO3)3·9H2O(分析纯,北京化工厂)溶解在蒸馏水中,室温下配制成0.2 mol·L-1的水溶液,搅拌条件(700 r·min-1)下滴加0.6 mol·L-1的Na2CO3(分析纯,国药集团化学试剂有限公司)水溶液,待成胶完全后将反应液静置过夜、抽滤、洗涤,所得白色沉淀110°C干燥12 h,在马弗炉中500°C焙烧6 h,研磨备用,记为Al2O3-2.
Al2O3载体的组成分析结果列于表1.
1.2 钌基氨合成催化剂的制备
采用浸渍法制备钌基氨合成催化剂,Ru负载的质量分数为5%.将载体用Ru3(CO)12(自制)的四氢呋喃(THF)(分析纯,天津化学试剂有限公司)溶液浸渍12 h,室温蒸去溶剂,于60°C真空干燥12 h,再在300°C下H2气氛加热3 h以分解Ru3(CO)12.所得样品分为两部分:一部分经压片、破碎、筛分后得无助剂的钌基催化剂Ru/Al2O3;另一部分用Ba(NO3)2(分析纯,上海化学工业专科学校实验工厂)的水溶液浸渍12 h,110°C干燥12 h,压片、破碎、筛分制备Ba:Ru摩尔比分别为1:1、3:1、5:1的Ba-Ru/Al2O3催化剂.
1.3 催化剂的氨合成活性评价
(2)方案设计有问题。方案设计是进行造价的前提,现阶段,由于设计人员担忧方案设计出现风险,对设计方案偏保守,过分考虑了方案的安全性,导致配套工程大而全,最后影响工程造价。另外,设计方案的错、漏、补问题多,在项目建设的实施阶段频繁进行工程变更,导致施工无法连续进行,既浪费成本又影响工期和质量,降低投资效益。这些问题都源于方案设计中没有建立起更加严格的监督机制,未对项目进行审核,这需要设计单位从维护业主的角度出发,设计出更科学的方案。
氨合成反应在固定床反应器中进行,催化剂(20-40目)用量2 mL,反应压力3.0 MPa,反应气为H2和N2混合气,空速5000 h-1.活性测试前催化剂在常压下用反应气在450°C活化6 h,然后在测试条件下至少稳定2 h,产物氨由气相色谱(Shimadzu GC-9A PORAPAK QS)在线分析.催化剂活性(x)由下式计算得到的单位时间内单位质量催化剂上生成的氨体积量(mL·g-1·h-1)表示.
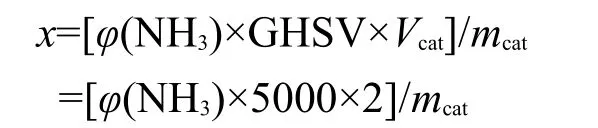
其中:mcat是催化剂质量,单位g;GHSV(gas hourly space velocity)是体积空速,单位h-1;Vcat是催化剂体积,单位mL.
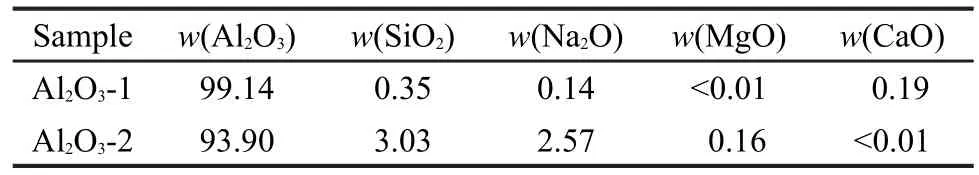
表1 Al2O3的组成(%)分析Table 1 Composite(%)analyses ofAl2O3samples
1.4 催化剂的表征
XRF在荷兰PANalytical公司的Magix PW2403 X射线荧光光谱仪上进行.XRD测试在荷兰PANalytical公司X'pert PRO型XRD衍射仪上进行,采用Cu Kα射线,管电压40 kV,管电流40 mA,2θ扫描范围为15°-85°,扫描速率0.2(°)·s-1.N2-低温物理吸附在ASAP 2010型气体吸附仪(USA Micromeritics)上于-196°C进行,测试前,样品在200°C真空条件下脱气处理5 h.样品的比表面积采用BET方程计算求得;孔容取p/p0=0.99时相应的吸附体积;孔径分布曲线由BJH公式计算得到.催化剂微观形貌由日本JEOL公司的JEM-2010型透射电子显微镜得到.测试前样品加入乙醇溶液中用超声波分散,在微栅上晾干后直接拍摄,电子加速电压200 kV,分辨率为 0.194 nm.H2-TPR测试采用先权公司TP-5080型全自动多用吸附仪.具体操作为:称取50 mg催化剂样品置于石英管中,200°C下高纯Ar吹扫30 min,降至室温,继续用高纯Ar吹扫至基线平稳.以10%H2-90%Ar混合气进行程序升温还原至700°C,还原气流速30 mL·min-1,升温速率10°C· min-1.利用NH3-TPD化学吸附法测定样品酸碱性.采用先权公司TP-5080型全自动多用吸附仪.具体操作为:称取100 mg催化剂样品于石英管中,在流速为20 mL·min-1的高纯Ar气流中升温至450°C,吹扫样品1 h以脱除吸附在样品表面的杂质,然后在Ar气流中降至室温,将Ar气流切换成高纯NH3气流,室温下吸附NH31 h,吸附完毕后切换回流速为20 mL·min-1的Ar气流,于120°C吹扫样品表面物理吸附的NH3和弱吸附的NH3后,以20°C·min-1的升温速率升温至850°C,使吸附在样品表面的NH3脱附.催化剂表面元素的化学组成和化学态用英国VG Scientific公司的ESCALAB 210型X射线光电子能谱仪(XPS)分析.使用Mg Kα射线(1253.6 eV,300 W),在大约5×10-9Pa下收集XPS样品.XPS结合能用污染碳C 1s(284.6 eV)进行校正,以便于催化剂与标准化合物的对比.
1.5 B5-Ru原子球棍模型绘制
采用FIZ/NIST Inorganic Crystal Structure Database Version 1.3.3软件搜索空间构型为P63/MMC的Ru元素-ICSD#43710,将文件保存为CIF格式.采用DIAMOND Crystal and Molecular Structure Visualization软件将此文件打开,绘制3×3×3 cells的 Ru0晶体.从中寻找满足以下两个条件的5个Ru原子的集合体:(a)暴露出三叠层空位(three-fold hollow site)和一个桥位(bridge site);(b)部分原子是低坐标的表面原子[23].
2 结果与讨论
2.1 催化剂的氨合成活性
表2为氧化铝负载钌基催化剂的比表面积和氨合成活性结果.可以看出,在不加入钡助剂时,Ru/ Al2O3-1和Ru/Al2O3-2的氨合成活性基本相同,在整个反应温度范围内均较低.随着BaO含量的增大, Ru/Al2O3-1的氨合成活性先迅速增大,Ba-Ru(3:1)/ Al2O3-1在450°C活性达到210.3 mL·g-1·h-1,继续增大助剂钡含量至n(Ba):n(Ru)=5:1时,活性急剧下降至Ru/Al2O3-1的水平.BaO助剂对Ru/Al2O3-2的催化活性的影响与此类似,不同的是,对于Al2O3-2载体,当n(Ba):n(Ru)=1:1时,在450°C时活性达到最大值221.2 mL·g-1·h-1,稍高于Ba-Ru(3:1)/Al2O3-1的活性.继续增大助剂钡含量至n(Ba):n(Ru)=5:1时,观察到活性下降至Ru/Al2O3-2的水平.这表明,存在一个最佳的BaO含量值,对表面性质不同的γ-Al2O3载体,这个最佳值也不同.张新波等[4]利用BaO改性γ-Al2O3载体,也发现氨合成活性随BaO含量的增大而增大,达到一个极大值后,继续再加入BaO,则造成活性的急剧下降.文献[15-16]的结果表明,BaO助剂的作用主要分为中和酸性部位和传输电子给Ru两种,当摩尔比n(Ba):n(Ru)为2-4时,活性随钡助剂含量的增大而呈上升趋势,此时适量的BaO助剂可以中和表面酸性和传递电子给Ru0,进一步增加BaO含量造成催化剂表面的Ru活性中心被BaO层覆盖,影响Ru0-载体界面在反应气氛中的充分暴露,导致活性的下降.
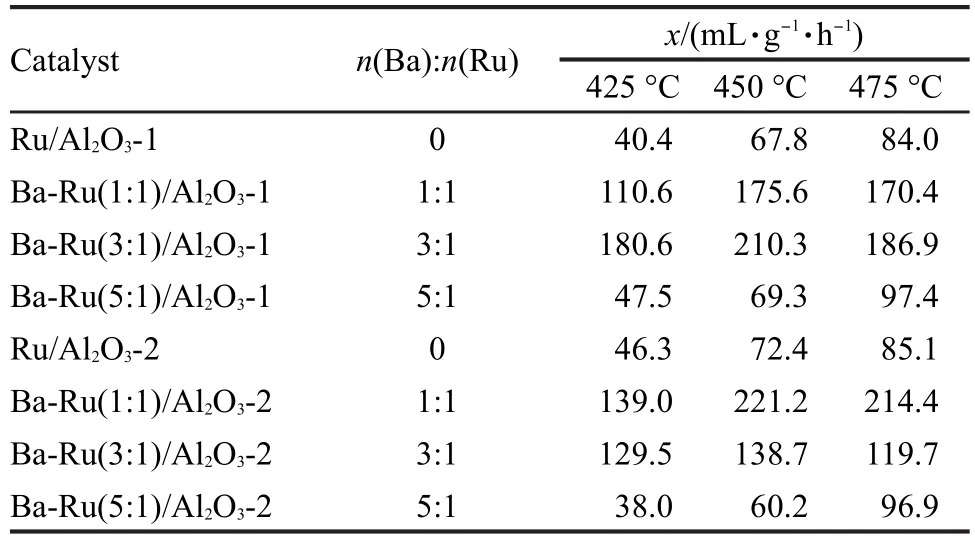
表2 Al2O3负载钌基催化剂的氨合成活性评价结果Table 2 Rate of ammonia synthesis over theAl2O3 supported Ru catalysts
2.2 XRD结果
图1为氧化铝负载钌基催化剂经H2于450°C还原后的XRD谱.可以看出,经H2还原处理后所有催化剂中均不存在Ru0或RuO2相,这可能是由于金属钌分散均匀或者以非晶形式存在[17].Aika等[10]认为,碱性助剂的加入可以增大钌活性物种的离子势,促进电子从Ru0到N2反键轨道的转移,从而降低N2解离吸附的活化能,而这一步正是氨合成反应的速率控制步骤.加入BaO助剂后,n(Ba):n(Ru)=1:1时,出现对应于BaCO3(JCPDS No.05-0378)相的新衍射峰,霍超等[18]认为这是由于经焙烧后生成的BaO晶粒较小,碱性较强,极易吸收空气中的CO2,从而生成BaCO3晶相.助剂钡含量增大至n(Ba): n(Ru)=3:1时,除了BaCO3的衍射峰,同时还检测到BaAl2O4(JCPDS No.17-0306)衍射峰.BaO在Al2O3表面的单层分散阈值在5%-10%(质量分数)之间,当BaO含量超过10%,BaO与Al2O3发生固相反应形成对应于BaAl2O4的新衍射峰[4].
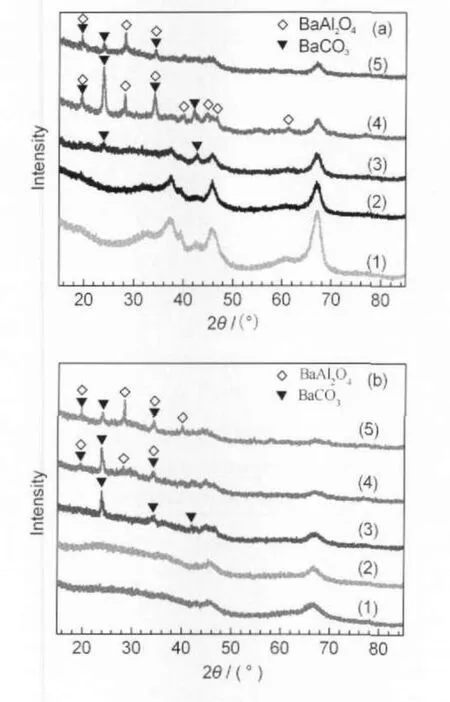
图1 Al2O3负载钌基催化剂的XRD谱图Fig.1 XRD patterns for theAl2O3supported Ru catalysts(a)Al2O3-1,(b)Al2O3-2;(1)Al2O3,(2)Ru/Al2O3,(3)Ba-Ru(1:1)/Al2O3, (4)Ba-Ru(3:1)/Al2O3,(5)Ba-Ru(5:1)/Al2O3
图1(b)可见,与Al2O3-1相同,沉淀法合成的Al2O3-2也为γ型,不过,尽管晶型相同,与Al2O3-1载体相比,Al2O3-2载体衍射峰较宽,表明Al2O3-2的晶粒尺寸明显要小于Al2O3-1的晶粒尺寸.在n(Ba): n(Ru)=1:1时,Ba-Ru(1:1)/Al2O3-2的BaCO3相衍射峰强度要强于Ba-Ru(1:1)/Al2O3-1的.在n(Ba):n(Ru)= 3:1时,Ba-Ru(3:1)/Al2O3-1的BaAl2O4和BaCO3相衍射峰强度均强于Ba-Ru(3:1)/Al2O3-2的.这可能是由于BaO与不同来源的Al2O3载体作用力的强弱不同所致.我们发现不同来源γ-Al2O3负载钌基催化剂的XRD谱图均有共同点,即BaO含量较低时,在催化剂中均不存在BaAl2O4相,而当BaO含量超过一定值后,出现了大量BaAl2O4相,对于不同来源的γ-Al2O3,BaAl2O4相的强度不同,这表明BaO与来源不同的γ-Al2O3的作用力存在差异.
2.3 N2低温物理吸附-脱附结果
表3列出了Al2O3载体及其负载钌基催化剂的比表面积、孔容和平均孔径结果.Al2O3-1负载的钌基催化剂的比表面积和孔体积均大于Al2O3-2负载的钌基催化剂的.随着助剂钡含量的增加,Ba-Ru/ Al2O3催化剂比表面积和孔体积均呈现逐渐减小的趋势.图2为不同样品的N2吸附-脱附等温线,Al2O3负载的钌催化剂具有典型的IV型等温线,表明其具有介孔结构,在相对压力p/p0为0.4-1.0范围内出现因N2的毛细管凝聚而产生的H2型滞后环,表明孔径分布相对较窄,孔形主要是蠕虫孔道[19-20].图3为BJH法从N2吸附-脱附等温线计算得到的Al2O3负载钌基催化剂孔径分布.由脱附支可得Ru/Al2O3-1、Ba-Ru(1:1)/Al2O3-1、Ba-Ru(3:1)/Al2O3-1和Ba-Ru(5: 1)/Al2O3-1催化剂最可几孔径分布分别位于3.2和6.6 nm、3.2和6.8 nm、3.5和6.7 nm、3.4和6.6 nm,而根据吸附支计算,孔径分布分别为3.5、3.8、5.5、6.1 nm,吸附支与脱附支计算的PSD结果差异表明材料中存在表面缺陷或特殊孔道结构,吸脱附过程中造成张力强度效应(tensile strength effect)[20-22].与Al2O3-1负载钌基催化剂类似,Ru/Al2O3-2、Ba-Ru(1: 1)/Al2O3-2、Ba-Ru(3:1)/Al2O3-2和Ba-Ru(5:1)/Al2O3-2催化剂由脱附支得到最可几孔径分布分别位于3.4、5.4、7.4 nm,3.5 nm,3.4、4.0 nm,3.3 nm,吸附支计算前3种催化剂的孔径分布分别为7.6、4.6、5.5 nm, Ba-Ru(5:1)/Al2O3-2催化剂孔道几乎全部堵塞,因此在吸附支上几乎看不到最可几孔径的分布,BaO助剂的过量加入对孔道的堵塞现象在一定程度上阻滞了Ru0-载体界面在反应气中的充分暴露.
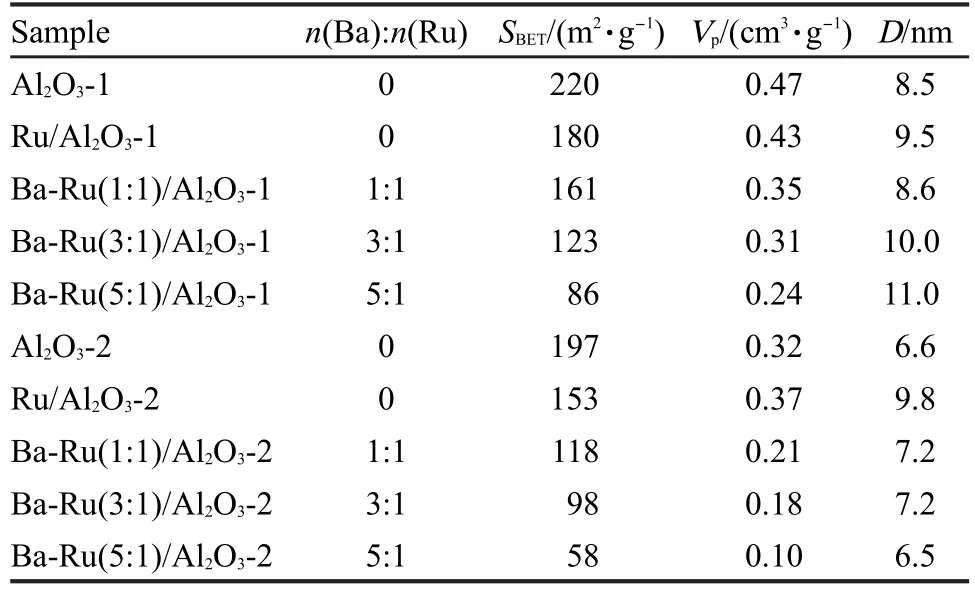
表3 载体和催化剂的织构性质Table 3 Pore texture properties ofAl2O3supports and catalysts
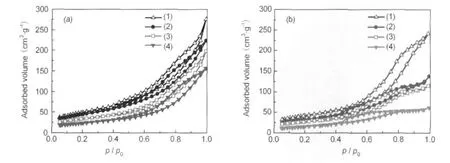
图2 Al2O3负载钌基催化剂的N2吸附-脱附等温线Fig.2 N2adsorption-desorption isotherms for the Al2O3supported Ru catalysts(a)Al2O3-1,(b)Al2O3-2;(1)Ru/Al2O3,(2)Ba-Ru(1:1)/Al2O3,(3)Ba-Ru(3:1)/Al2O3,(4)Ba-Ru(5:1)/Al2O3
2.4 TEM结果
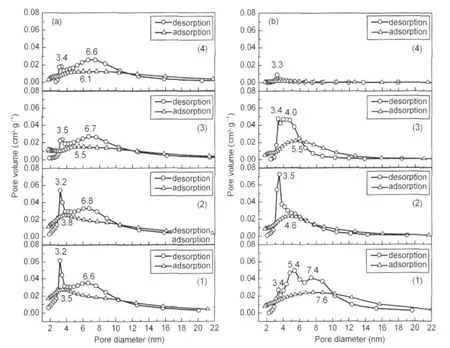
图3 Al2O3负载钌基催化剂的孔径分布图Fig.3 Pore size distribution of theAl2O3supported Ru catalysts(a)Al2O3-1,(b)Al2O3-2;(1)Ru/Al2O3,(2)Ba-Ru(1:1)/Al2O3,(3)Ba-Ru(3:1)/Al2O3,(4)Ba-Ru(5:1)/Al2O3
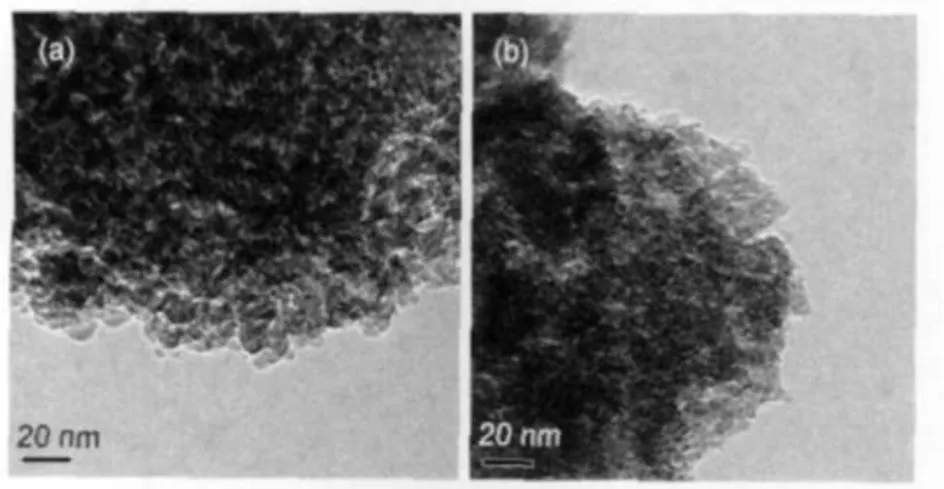
图4 Ba-Ru(1:1)/Al2O3催化剂的TEM照片Fig.4 TEM images of Ba-Ru(1:1)/Al2O3catalysts(a)Ba-Ru(1:1)/Al2O3-1;(b)Ba-Ru(1:1)/Al2O3-2
图4为于450°C用H2还原后的Ba-Ru(1:1)/ Al2O3-1和Ba-Ru(1:1)/Al2O3-2催化剂的TEM照片. Al2O3-1和Al2O3-2均存在大量蠕虫状孔道,与N2低温物理吸附的结果一致(图2).Ru颗粒比较均匀地分散在Al2O3载体上,Ru3(CO)12/Al2O3分散度比较高的原因是由于Ru3(CO)12和Al2O3表面的羟基反应,生成了单原子层锚式络合物[16].随机选取100个Ru颗粒进行粒径分布统计,结果表明,Ru粒径均为1-2 nm.钌基催化剂表面上进行的氨合成反应是典型的结构敏感反应,密度泛函理论(DFT)计算表明,对N2的解离和氨合成反应最具有活性的位置是满足两个条件的5个Ru原子的集合体(如1.5部分所述)[2].B5位的数量与Ru粒子大小有密切关系(图5), Jacobsen等[23]认为Ru粒径在1.5-2.0 nm时,较易形成B5结构反应活性中心,有利于活性的提高.随着Ru晶粒的长大,低坐标表面原子所占比率会减少, B5中心数量下降.Al2O3负载钌基催化剂在形成B5活性中心方面是有利的,而其活性的差异则主要是由其体系的电子性质影响所致.
2.5 H2-TPR结果

图5 Ru晶体中N2吸附解离的B5位示意图Fig.5 Ball model illustrating B5 sites on the side facets of Ru crystallites
图6(1-4)为Al2O3-1负载钌基催化剂的TPR谱. Ru/Al2O3-1催化剂呈现典型的“双峰”还原过程.研究表明,钌催化剂上存在两种Ru物种的还原峰,低温峰为呈现高分散状态的Ru粒子或表相的Ru物种-RuOx的还原峰,而高温峰为体相Ru粒子-RuO2的还原峰,前者的峰温较低(150-180°C),而后者的还原温度较高(300°C)[24].Ru/Al2O3-1催化剂的Ru物种在92和325°C分别发生还原;Ba-Ru(1:1)/Al2O3-1催化剂的Ru物种在25-125°C的范围内几乎已经被全部还原,在175-475°C的温度范围出现宽的高温还原峰,强度较小.这表明适量BaO助剂的加入促进了钌催化剂的还原表现,n(Ba):n(Ru)=1:1时,助剂BaO在载体表面呈单分散状态,Ru-BaO界面在反应气氛中便于暴露,Ru物种还原容易.Ba-Ru(3: 1)/Al2O3-1催化剂在整个还原温度范围内出现三个还原峰,峰温在140和312°C的还原峰分别为Ru物种的低温、高温还原峰,与不加助剂的Ru/Al2O3-1相比,过量助剂BaO的加入使Ru物种的还原变得更加困难[25],这是由于当n(Ba):n(Ru)为3:1和5:1时,助剂量超过了单层分布的量,造成大量BaO助剂附着在Ru和载体表面,Ru-BaO界面不能在反应气氛中暴露,造成其还原困难,还原峰温度上升,过量的BaO助剂阻滞了Ru物种的还原过程[2].Ba-Ru(3:1)/ Al2O3-1催化剂的第三个宽的肩峰出现在400-550°C温度区间,XRD结果(图1a(4))表明Ba-Ru(3:1)/ Al2O3-1催化剂中存在大量BaCO3物相,因此,这个还原峰是由于BaO助剂与空气作用生成的BaCO3的热分解造成.BaCO3是碱土金属碳酸盐中最稳定的碳酸盐,在空气中的理论分解温度达到1287°C.霍超等[26]系统研究了氢气还原气氛下催化剂中BaCO3的热稳定性,他们认为,在Ru0表面存在氢溢流现象,H2在Ru作用下解离为氢原子,从Ru0表面溢流至载体表面,从而有效地促进了BaCO3的低温分解过程.BaO是钌基氨合成催化剂的有效助剂, Hansen等[27]认为靠近于Ru晶体B5活性位的BaO是催化剂的电子助剂,可以极大地促进氨合成活性.氨合成活性结果也表明,Ba-Ru(3:1)/Al2O3-1催化剂在450°C,5 MPa,5000 h-1反应条件下活性达到210.3 mL·g-1·h-1,明显高于其他Al2O3-1负载钌基催化剂的氨合成活性.Ba-Ru(5:1)/Al2O3-1样品在282°C出现较强的体相RuO2还原峰,再次证实BaO助剂的过量加入确实对Ru物种的还原过程有阻滞作用.
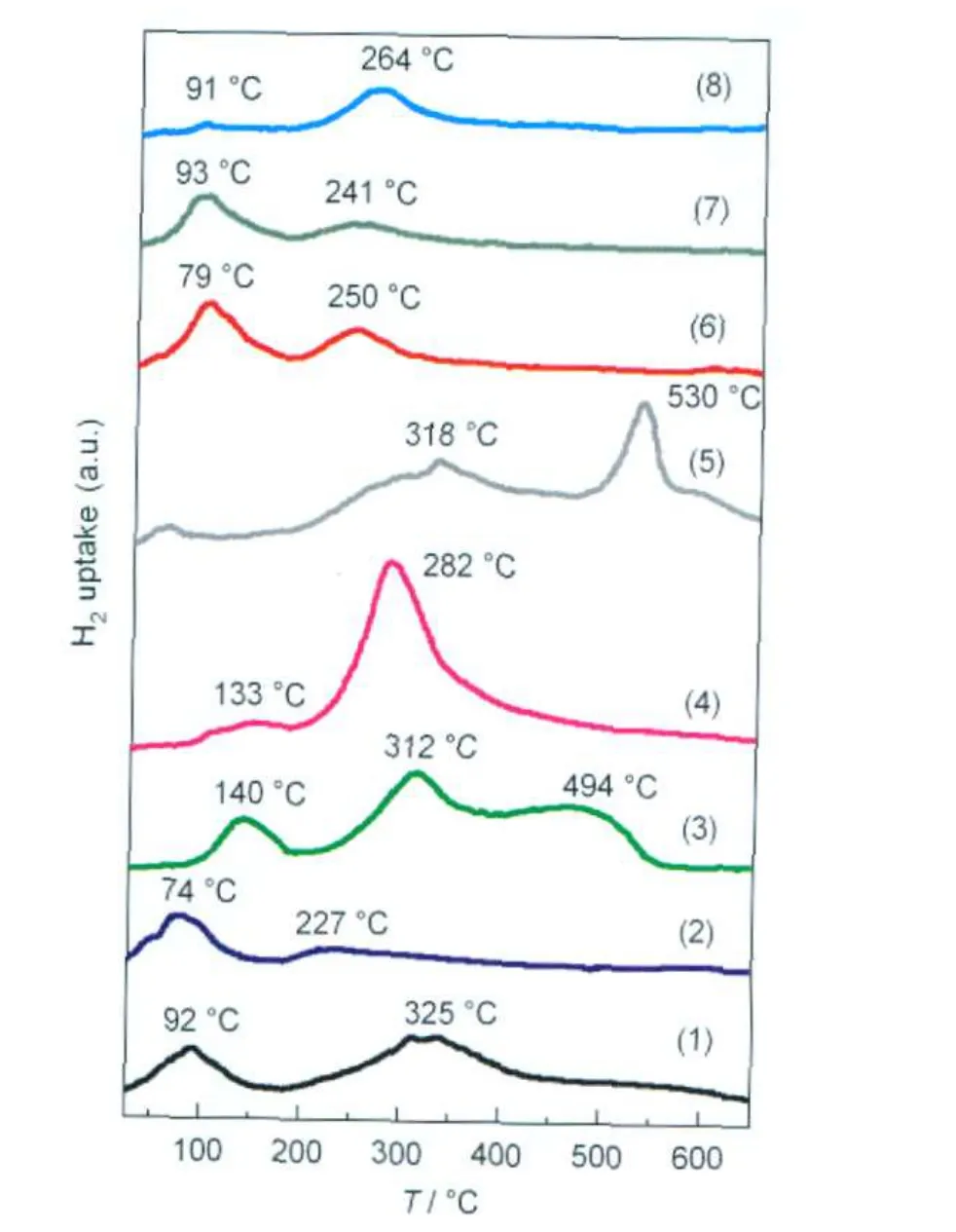
图6 Al2O3负载钌基催化剂的H2-TPR谱Fig.6 H2-TPR patterns of theAl2O3supported Ru catalysts(1)Ru/Al2O3-1;(2)Ba-Ru(1:1)/Al2O3-1;(3)Ba-Ru(3:1)/Al2O3-1; (4)Ba-Ru(5:1)/Al2O3-1;(5)Ru/Al2O3-2;(6)Ba-Ru(1:1)/Al2O3-2; (7)Ba-Ru(3:1)/Al2O3-2;(8)Ba-Ru(5:1)/Al2O3-2
图6(5-8)为Al2O3-2负载钌基催化剂的TPR谱.与Ru/Al2O3-1相同,Ru/Al2O3-2样品的还原过程也分为两步还原,然而,很明显其还原峰温度(318和530°C)要高于前者,这可能是由于Al2O3-2与Ru物种的作用程度较Al2O3-1更强,造成还原温度升高.与Al2O3-1负载钌基催化剂的还原表现不尽相同, n(Ba):n(Ru)为1:1和3:1时,BaO助剂的加入造成Ba-Ru/Al2O3-2的还原峰向低温方向移动,Ba-Ru(1: 1)/Al2O3-2峰温分别为79和250°C,Ba-Ru(3:1)/ Al2O3-2峰温分别为93和241°C,且大部分均在低温时被还原.这表明BaO在Al2O3-2上单分散分布的量要大于在Al2O3-1上的.n(Ba):n(Ru)=5:1时,Ba-Ru(5: 1)/Al2O3-2峰温分别为91和264°C,且大部分在高温时被还原,这表明此时BaO在催化剂表面并非单分散状态,而是覆盖在Ru物种和载体表面,造成Ru-BaO界面难于接触反应气,还原困难.活性评价结果表明Ba-Ru(1:1)/Al2O3-2催化剂的活性(450°C, 221.2 mL·g-1·h-1)稍高于Ba-Ru(3:1)/Al2O3-2的(450°C,138.7 mL·g-1·h-1)的活性,这表明钌基催化剂的氨合成活性与Ru物种的还原性质密切相关,不仅是表相RuOx物种的还原,体相RuO2的还原也对反应有影响.Ba-Ru(5:1)/Al2O3-2的钌物种主要以体相RuO2形式被还原,氨合成活性较低.Dahl等[28]认为B5位是对N2的解离和氨合成反应最具有活性的Ru原子排布方式,而B5位的丰度则取决于Ru粒子的形状和尺寸大小,Ru粒径与还原温度相关,因此, Ru物种的还原性质对催化剂的氨合成活性有较大影响.
2.6 NH3-TPD结果
在氨合成反应中,N≡N键的断裂是非常困难的,碱金属或碱土金属等供电子助催化剂的加入能够提供电子给活性组分Ru,削弱N2分子内的键能,增强N2的解离吸附,从而提高氨合成反应速度.氨合成活性与载体的碱性成正比,即碱性越大,活性越高[23,25].Al2O3是一种常用的催化剂载体,是强的Lewis酸,室温下Al2O3表面为OH-所覆盖,呈非酸性,当温度升高时,表面的OH-与相邻的H生成H2O分子而被除去,形成不饱和配位的Al3+,成为Lewis酸中心,一般在>300°C才能观察到酸性,而Lewis酸吸附水气后形成具有弱Brønsted特征的质子酸部位[29].除了酸特征外,脱水过程中形成的带负电荷的O-形成了碱性部位.由于酸性部位阻滞了氨合成反应过程中载体或者助剂到Ru的电子传输,因此,添加碱性助剂是必要的.我们利用NH3-TPD考察了纯氧化铝载体和添加碱性助剂BaO的Ba-Ru/Al2O3催化剂的表面酸性性质.图7为Al2O3载体的NH3-TPD谱图,Al2O3-1在整个NH3脱附温度范围内明显出现两个峰,峰顶温度分别为167和568°C,表明在氧化铝表面明显存在弱酸性位和强酸性位.脱附曲线在从低温到高温过程中没有回到基线,表明表面存在一定数量的中等强度的酸性位[30].Somorjai等[31]认为Al2O3表面存在3类不同配位数的Al原子:高配位数Al原子(低表面能,弱酸中心)、中等配位数Al原子(中等表面能,中等酸中心)和低配位数Al原子(高表面能,强酸中心).OH-分别与酸性不同的酸中心结合,从而形成三类酸强度各不相同的表面—OH.
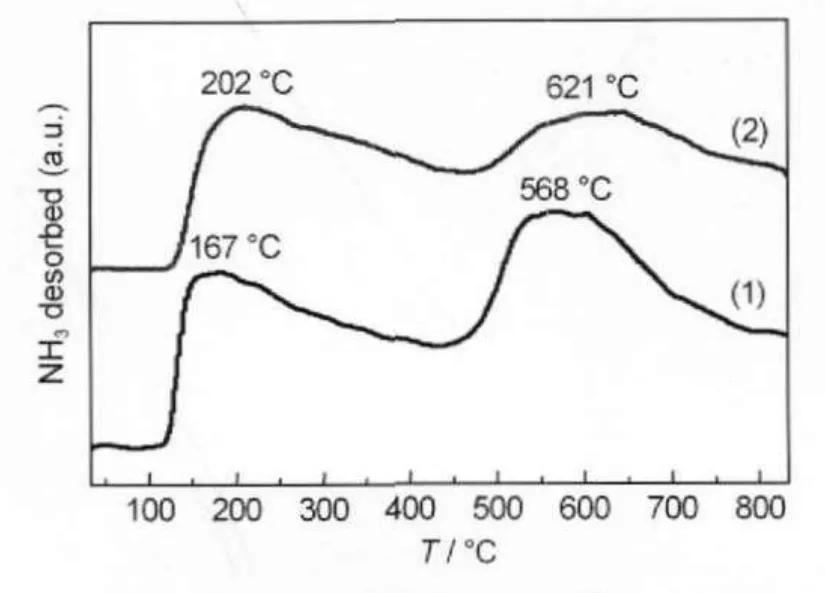
图7 Al2O3的NH3-TPD谱Fig.7 NH3-TPD spectra ofAl2O3 (1)Al2O3-1;(2)Al2O3-2
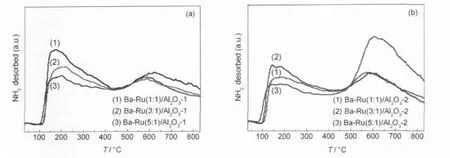
图8 Al2O3负载钌基催化剂的NH3-TPD谱Fig.8 NH3-TPD spectra of theAl2O3supported Ru catalysts (a)Ba-Ru/Al2O3-1;(b)Ba-Ru/Al2O3-2
以NH3为吸附质的Al2O3的TPD谱图,虽然因为Al2O3的制备方法不同而呈现不尽相同的谱图,但其峰形弥散又相互重叠则是共同的特点,表明酸强度分布并不均匀.与Al2O3-1相比,Al2O3-2的NH3-TPD谱图轮廓相同,不同的是其脱附峰峰顶温度(202和621°C)较前者更高.这表明沉淀法合成的Al2O3-2的表面酸强度较Al2O3-1更强.NH3-TPD峰面积的分析数据表明,Al2O3-2的Lewis酸性位数量明显比Al2O3-1的多.我们考察了BaO助剂添加量对不同来源Ba-Ru/Al2O3体系酸性的影响(图8),BaO助剂在Ba-Ru/Al2O3-1体系中存在状态随添加量而不同, n(Ba):n(Ru)=1:1时,主要以BaCO3形态存在;n(Ba): n(Ru)为3:1和5:1时,主要以BaCO3和BaAl2O4形态存在.由图8(a)可以看出,体系弱酸性位数量随助剂的添加量增大而减小,体系总酸性位数量也是逐渐减少,这表明BaO助剂的添加造成体系酸性下降. BaO助剂添加量对Ba-Ru/Al2O3-2体系酸性的影响则不相同,体系弱酸性位数量先增加后减少,在n(Ba):n(Ru)=3:1时达到极大值,而强酸性位数量则在n(Ba):n(Ru)=5:1时达到极大值(图8(b)).BaO助剂添加的结果是体系总酸性位数量增大,n(Ba):n(Ru)= 5:1时总酸性达到极大值.BaO的加入没有降低体系酸性,相反,体系酸性随BaO的加入而增大,这种现象可能是由于Ru/Al2O3催化剂对NH3的催化分解作用所致,Nagai等[32]认为,Ru/Al2O3样品的NH3-TPD高温峰归因为NH3分解成N2和H2,而对Ru的不同处理过程会造成NH3分解作用的强弱区别,这可能就是我们在Ba-Ru/Al2O3-1体系中并没有观察这种现象的原因.图9为氨合成活性与体系总酸量随助剂添加量的变化曲线,对Ba-Ru/Al2O3-1体系来说,活性随助剂BaO添加量呈“火山”曲线,而总酸性则单调减小,这可能是由于体系酸性、比表面积和氨合成活性之间存在最优比例(图9(a)).Ba-Ru/Al2O3-2体系氨合成活性与体系总酸性成反比(图9(b)),这表明对于Al2O3-2负载钌基催化剂,氨合成活性与体系碱性是成正比的,体系碱性对氨合成活性的影响要强于比表面对其的影响.
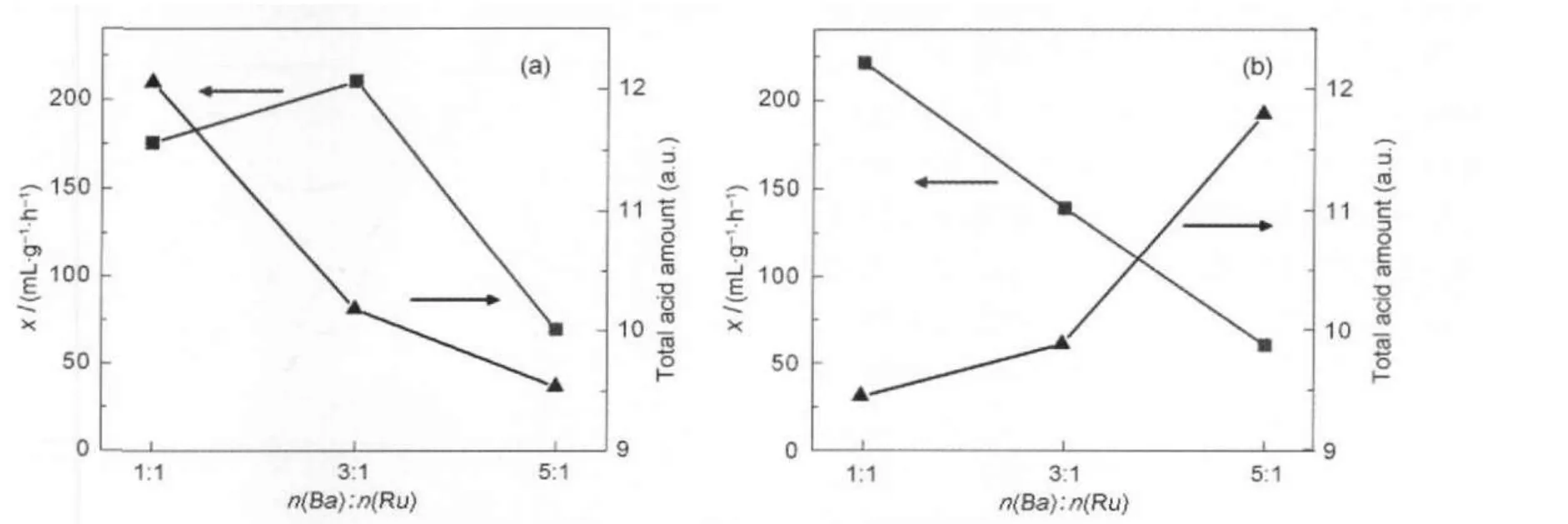
图9 Ba-Ru/Al2O3催化剂氨合成活性与酸总量随BaO助剂添加量的变化曲线Fig.9 Curves of ammonia formation rate and total acid amount of Ba-Ru/Al2O3vsBa/Ru molar ratio(a)Ba-Ru/Al2O3-1;(b)Ba-Ru/Al2O3-2
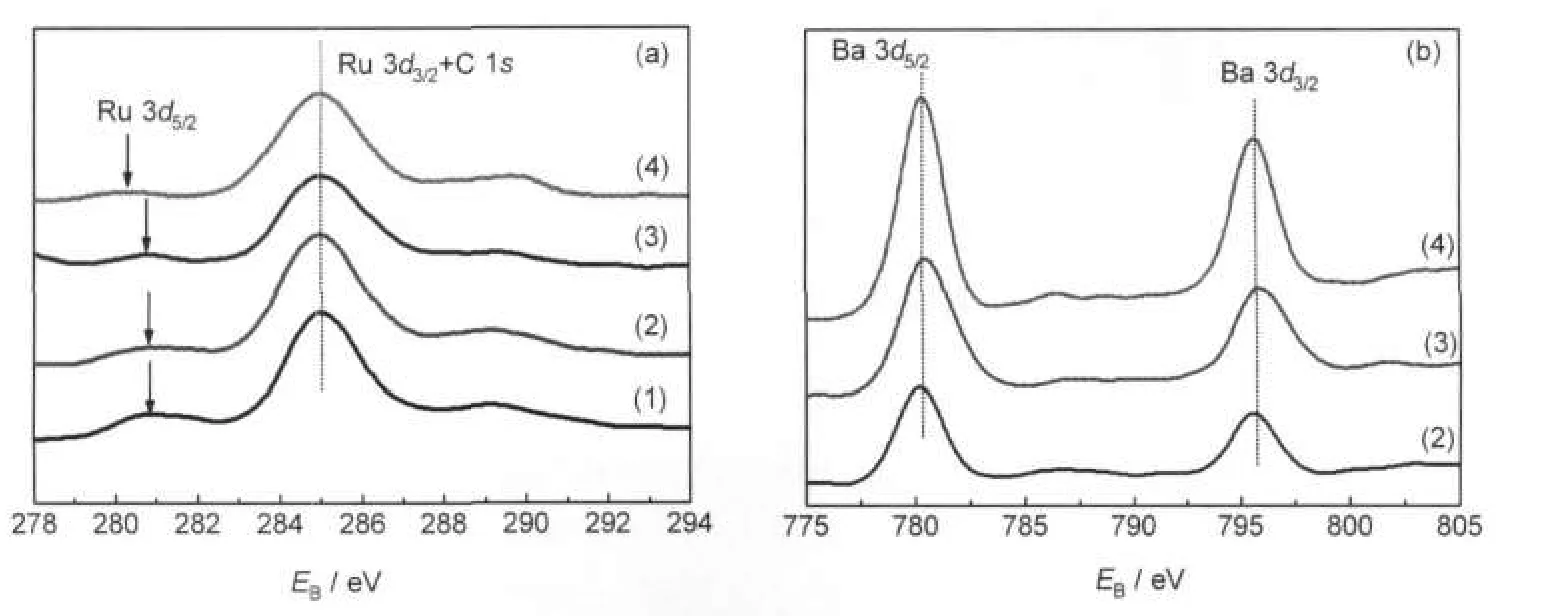
图10 Ba-Ru/Al2O3-2催化剂的Ru 3d和Ba 3d XPS谱Fig.10 XPS patterns of Ru 3d and Ba 3d for Ba-Ru/Al2O3-2 catalysts(a)Ru 3d,(b)Ba 3d;(1)Ru/Al2O3-2,(2)Ba-Ru(1:1)/Al2O3-2,(3)Ba-Ru(3:1)/Al2O3-2,(4)Ba-Ru(5:1)/Al2O3-2
2.7 XPS结果
图10为Ba-Ru/Al2O3-2催化剂的XPS谱图.由于轨道-耦合作用,Ru的谱图包含3d3/2和3d5/2两个峰,Ru 3d3/2与C 1s峰重叠,碳污染来自催化剂表面与空气中CO2的化学吸附或者表面键合作用,因此,一般以Ru 3d5/2峰为研究对象[9].如图10(a)所示, Ru/Al2O3催化剂中Ru 3d5/2峰位于280.7 eV,随着BaO助剂含量的增加,Ru 3d5/2峰位依次为280.7、280.7和280.2 eV.Ru03d5/2为280.0 eV,RuO23d5/2为280.6 eV,显然,Ru/Al2O3、Ba-Ru(1:1)/Al2O3和Ba-Ru(3:1)/ Al2O3催化剂中钌物种均以RuO2形态存在,这是由于样品暴露在空气中表面被氧化的原因.而对于Ba-Ru(5:1)/Al2O3催化剂,其Ru 3d5/2比RuO2低0.4 eV,比Ru0高0.2 eV,钌物种极有可能以Ru0形态存在[33],我们认为其原因可能是由于加入的BaO助剂量较大,大量BaO包裹在Ru粒子的表面,BaO作为电子助剂倾向于将电子传递给钌物种,从而在一定程度上抑制了RuO2的形成.然而,这种大剂量的包覆作用也同时造成BaO-Ru界面难于暴露在反应气氛中,造成活性的下降(表2).图10(b)为Ba-Ru/ Al2O3-2催化剂体系的钡助剂的XPS谱图,Ba-Ru(1: 1)/Al2O3、Ba-Ru(3:1)/Al2O3和Ba-Ru(5:1)/Al2O3中的Ba 3d5/2分别位于780.2、780.4和780.3 eV,基本没有变化,XRD谱图表明Ba-Ru(1:1)/Al2O3中钡物相只有BaCO3,而Ba-Ru(3:1)/Al2O3和Ba-Ru(5:1)/Al2O3中则同时存在BaCO3和BaAl2O4物相.
3 结论
载体的物理性质,如结构、织构性质影响了Ru粒子在其表面的分散状态以及尺寸;载体的化学性质,如表面官能团、表面酸碱性质影响了Ru物种的还原性质和电子密度.BaO助剂添加量不同导致BaO与γ-Al2O3的作用力不同,BaO与催化剂体系钌物种的作用程度不同,从而进一步影响催化剂的氨合成活性,适量BaO的加入能够极大促进反应活性,此时,BaO同时起到促进钌物种的还原以及调节体系碱性的作用,过量的BaO的加入则会由于这两种作用强弱的失调而降低催化剂活性.对于不同来源的γ-Al2O3,这种最佳量也是不相同的,3 MPa, 5000 h-1,Ba-Ru(3:1)/Al2O3-1催化剂在450°C活性为210.3 mL·g-1·h-1,而相同反应条件下Ba-Ru(1:1)/ Al2O3-2在助剂含量远小于前者情况下达到221 mL· g-1·h-1.
1 Liang,C.H.;Wei,Z.B.;Xin,Q.;Li,C.Appl.Catal.A,2001, 208:193
2 Yang,X.L.;Hu,B.;Xia,C.G.;Xiong,X.M.;Mu,X.Y.Ind. Catal.,2010,18:5 [杨晓龙,胡 斌,夏春谷,熊绪茂,慕新元.工业催化,2010,18:5]
3 You,Z.X.;Inazu,K.;Aika,K.;Baba,T.J.Catal.,2007,251:321
4 Zhang,X.B.;Li,X.N.;Huo,C.;Cen,Y.Q.;Zhu,Y.F.;Liu,H. Z.Chin.J.Catal.,2002,23:207 [张新波,李小年,霍 超,岑亚青,祝一峰,刘化章.催化学报,2002,23:207]
5 Szmigiel,D.;Raróg-Pilecka,W.;MiŚkiewicz,E.;Gliński,M.; Kielak,M.;Kaszkur,Z.;Kowalczyk,Z.Appl.Catal.A,2004, 273:105
6 Xu,Q.C.;Lin,J.D.;Fu,X.Z.;Liao,D.W.Catal.Commun., 2008,9:1214
7 Ni,J.;Wang,R.;Lin,J.X.;Wei,K.M.Acta Phys.-Chim.Sin., 2009,25(3):519 [倪 军,王 榕,林建新,魏可镁.物理化学学报,2009,25(3):519]
8 Luo,X.J.;Wang,R.;Ni,J.;Lin,J.X.;Lin,B.Y.;Xu,X.M.;Wei, K.M.Catal.Lett.,2009,133:382
9 Larichev,Y.V.;Moroz,B.L.;Zaikovskii,V.L.;Yunusov,S.M.; Kalyuzhnaya,E.S.;Shur,V.B.;Bukhtiyarov,V.I.J.Phys.Chem. C,2007,111:9427
10 Miyazaki,A.;Balint,I.;Aika,K.;Nakano,Y.J.Catal.,2001,204: 364
11 Kadowaki,Y.;Aika,K.J.Catal.,1996,161:178
12 Siporin,S.E.;Davis,R.J.J.Catal.,2004,225:359
13 Wang,R.;Wei,K.M.;Lin,J.X.;Yu,X.J.;Mao,S.L.Ind.Catal., 2005,13:31 [王 榕,魏可镁,林建新,俞秀金,毛树禄.工业催化,2005,13:31]
14 Mao,S.L.;Wang,R.;Yu,X.J.;Lin,J.X.;Wei,K.M.Chin.Rare Earth,2004,25:18 [毛树禄,王 榕,俞秀金,林建新,魏可镁.稀土,2004,25:18]
15 Fu,W.J.The Preparation and study of the ruthenium catalysts for ammonia synthesis[D].Fuzhou:Fuzhou University,2003 [傅武俊.钌催化剂的制备及氨合成催化性能的研究[D].福州:福州大学,2003]
16 Liu,H.Z.Ammoina synthesis catalysis:practice and theory. Beijing:Chemical Industry Press,2007:309,293 [刘化章.氨合成催化剂实践与理论.北京:化学工业出版社,2007:293,309]
17 Okal,J.Catal.Commun.,2010,11:508
18 Huo,C.;Yan,G.;Zheng,Y.F.;Yu,F.W.;Liu,H.Z.Chin.J. Catal.,2007,28:484[霍 超,晏 刚,郑遗凡,于凤文,刘化章.催化学报,2007,28:484]
19 Sharma,L.D.;Kumar,M.;Saxena,A.K.;Chand,M.;Gupta,J. K.J.Mol.Catal.A,2002,185:135
20 Li,Y.;Yang,Q.H.;Yang,J.;Li,C.Microporous Mesoporous Mat.,2006,91:85
21 Yuan,Z.Y.;Wang,J.Z.;Zhang,Z.L.;Chen,T.H.;Li,H.X. Microporous Mesoporous Mat.,2001,43:227
22 Groen,J.C.;Pérez-Ramírez,J.Appl.Catal.A,2004,268:121
23 Jacobsen,C.J.H.;Dahl,S.;Hansen,P.L.;Törnqvist,E.;Jensen, L.;Topsøe,H.;Prip,D.V.;Møenshaug,P.B.;Chorkendorff,I. J.Mol.Catal.A,2000,163:19
24 Seetharamulu,P.;Siva Kumar,V.;Padmasri,A.H.;David Raju, B.;Rama Rao,K.S.J.Mol.Catal.A,2007,263:253
25 Seetharamulu,P.;Hari Prasad Reddy,K.;Padmasri,A.H.;Rama Rao,K.S.;David Raju,B.Catal.Today,2009,141:94
26 Huo,C.;Xia,Q.H.;Luo,Y.;Yang,X.Z.;Liu,H.Z.Chin.J. Catal.,2009,30:537 [霍 超,夏庆华,骆 燕,杨霞珍,刘化章.催化学报,2009,30:537]
27 Hanse,T.W.;Wagner,J.B.;Hansen,P.L.;Dahl,S.;Topsøe,H.; Jacobsen,C.J.H.Science,2001,294:1508
28 Dahl,S.;Logadottir,A.;Jacobsen,C.J.H.;Nørskov,J.K.Appl. Catal.A,2001,222:19
29 Zhao,Y.Ind.Catal.,2002,10:54 [赵 琰,工业催化,2002,10: 54]
30 Walker,G.S.;Pyke,D.R.;Werrett,C.R.;Williams,E.; Bhattacharya,A.K.Appl.Surf.Sci.,1999,147:228
31 Somorjai,G.A.Science,1978,201:489
32 Nagai,M.;Koizumi,K.;Omi,S.Catal.Today,1997,35:393
33 Elmasides,C.;Kondarides,D.I.,Grünert,W.;Verykios,X.E. J.Phys.Chem.B,1999,103:5227
September 1,2010;Revised:September 29,2010;Published on Web:November 8,2010.
Effect of Alumina Support and Barium Oxide on the Structure and Catalytic Activity of Ruthenium Catalysts for Ammonia Synthesis
YANG Xiao-Long1,2XIA Chun-Gu1TANG Li-Ping1,2XIONG Xu-Mao1MU Xin-Yuan1HU Bin1,*
(1Lanzhou Institute of Chemical Physics,Chinese Academy of Sciences,Lanzhou 730000,P.R.China;2Graduate University of Chinese Academy of Sciences,Beijing 100049,P.R.China)
A series of Ba-Ru/Al2O3catalysts were prepared by the impregnation method using industrial alumina(Al2O3-1)and synthesized alumina(Al2O3-2)as supports.The catalysts were characterized by X-ray diffraction,N2adsorption-desorption,X-ray fluorescence spectroscopy,transmission electron microscopy,H2temperature-programmed reduction,NH3temperature-programmed desorption,and X-ray photoelectron spectroscopy.The effect of Al2O3and the BaO promoter on the phase structure,texture properties,morphology,surface properties,and catalytic activity in ammonia synthesis were investigated. The results indicate that the physical and chemical properties of Al2O3have a strong impact on the structure and activity of the ruthenium catalysts.The BaO promoter has a strong impact on the ruthenium catalyst in two ways:first,the amount of BaO added leads to a difference in the interaction between BaO and γ-Al2O3,which further influences the specific area and the porous structure of the catalysts;second, the addition of BaO influences the reduction process and the surface acidity and alkaline properties of the ruthenium catalysts.A proper amount of BaO promotes the activity and the optimal amount of BaO depends on the properties of the supports.
Ammonia synthesis;γ-Al2O3;Support;Ruthenium;Reduce;Acidity
O643.3;TQ113.2
∗Corresponding author.Email:hcom@lzb.ac.cn;Tel:+86-931-4968258.
The project was supported by the National Science Foundation for Distinguished Young Scholars of China(20625308).国家杰出青年科学基金(20625308)资助项目
猜你喜欢
今日农业(2020年20期)2020-11-26
福建基础教育研究(2019年8期)2019-05-28
科学大众(中学)(2019年3期)2019-05-17
汽车观察(2018年10期)2018-11-06
中学生数理化·八年级物理人教版(2017年6期)2017-11-09
科技知识动漫(2017年1期)2017-02-06
中国中西医结合皮肤性病学杂志(2016年4期)2016-07-18
当代化工研究(2016年7期)2016-03-20
橡胶工业(2015年8期)2015-07-29
少儿科学周刊·少年版(2015年1期)2015-07-07
