
α-MnO2负载纳米Au催化剂低温催化氧化CO和苯的性能
2011-12-21 06:32霍飞飞闫立娜程水源康天放
物理化学学报 2011年12期
叶 青 霍飞飞 闫立娜 王 娟 程水源 康天放
(北京工业大学环境与能源工程学院,北京100124)
α-MnO2负载纳米Au催化剂低温催化氧化CO和苯的性能
叶 青*霍飞飞 闫立娜 王 娟 程水源 康天放
(北京工业大学环境与能源工程学院,北京100124)
以尿素为沉淀剂用沉积-沉淀法制备了α-MnO2负载Au催化剂xAu/α-MnO2(x=0-7(对应的Au负载量(质量分数)分别为0-7%)),使用X射线粉末衍射(XRD)、N2-吸附/脱附、透射电镜(TEM)、X射线光电子能谱(XPS)和H2-程序升温还原(H2-TPR)等技术对所制样品进行了表征,并测定其对CO和苯的催化氧化性能.XRD结果表明,负载Au对α-MnO2载体结构影响不大,随Au含量的增加,Au颗粒明显增大.N2-吸附/脱附和TEM结果表明,Au的加入对xAu/α-MnO2的比表面积、孔容和孔径等结构性能影响较小,表明Au分布在α-MnO2载体表面,未阻塞其孔道.XPS结果表明,随着Au负载量的增加,xAu/α-MnO2中的O2-/或O-)、Mn4+/Mn3+和Au3+/ Au0的摩尔比在增加,表明其晶格氧、Mn4+和Au3+的浓度在增加.由于贵金属的溢氢作用,Au明显促进xAu/ α-MnO2氧化还原能力,其中3Au/α-MnO2具有最高的氧化还原性.负载Au明显影响xAu/α-MnO2样品的催化活性,xAu/α-MnO2的催化性能与Au的分散性、氧化还原性能及表面氧物种的种类密切相关,其中3Au/α-MnO2显示出最佳活性,其催化氧化CO的T90=80°C,苯的T90=200°C.
α-MnO2负载Au催化剂;低温还原性;相互作用;CO氧化;苯催化燃烧
1 引言
CO的催化氧化已经在很多领域得到广泛的应用,如封闭式内循环CO2激光器、CO气体探测器、室内空气净化以及密闭空间微量CO的消除等领域.1近年来,在富H2气氛下CO的优先催化氧化(COPROX)也得到广泛的研究和应用.2此外,由于CO催化氧化反应的简单性,其常被作为催化研究中的探针反应之一.3因此,CO催化氧化反应具有较重要的理论意义和实际应用价值.
挥发性有机物(VOCs)大多来自于移动源和工厂排放的废气,其对大气环境和人体健康危害极大,所以消除VOCs气体污染具有重要的意义.通常有机物的消除是在高温(>1000°C)条件下对其进行燃烧处理,能源消耗较大.催化氧化技术通常被认为是最有效的处理VOCs的技术,其具有操作温度低(<500°C)、能源消耗低、效率高和操作费用低等优点,极具可行性,4但高效的催化剂是此技术的关键.
近年来,人们制备了大量的催化氧化VOCs气体的催化剂,包括贵金属(Pt、Pd、Rh、Au)负载型催化剂和过渡金属(Cr、Co、Cu、Ni、Mn)氧化物负载型催化剂.5其中,贵金属负载型催化剂在低温具有较高的活性,但是高昂的价格及在使用过程中容易烧结和中毒限制了它们的使用.6因此急需开发一种具有高的热稳定性和抗中毒能力的廉价催化剂.
体相/或氧化锰负载的催化剂常常被用做CO、甲烷和碳氢化合物催化氧化的催化剂.7通常一种金属氧化物负载催化剂常常很难与贵金属催化剂相媲美,而将两种或更多的金属氧化物结合制备的复合催化剂可以获得较高的催化活性.如Mn-Fe复合氧化物催化剂比沸石负载Pt催化剂具有更高的催化氧化含氧有机物的性能,8我们前期研究表明,将少量的Ag负载在氧化锰上制备的催化剂(Ag/ MnO2)具有较好的催化氧化CO和VOCs气体的活性.9
对于Au负载型催化剂,自从1987年Haruta等10报道Au负载型催化剂对CO低温催化氧化具有很高的活性以来,人们对纳米金负载型催化剂的CO催化氧化性质进行了广泛的研究.金负载型催化剂的催化性能与Au颗粒大小、载体性质和催化剂的制备方法密切相关.11目前人们普遍认为,高度分散态纳米金对低温催化氧化CO反应具有极好的催化活性.12当Au高度分散在金属氧化物(例如TiO2、 α-Fe2O3和Co3O4)上时,可以获得高的催化氧化活性.13
本研究基于纳米Au和氧化锰的催化氧化特性,将Au与氧化锰组成复合催化剂,研究其结构和催化氧化CO和VOCs的特性,其中苯作为代表VOCs的模型气体,以期获得其结构与催化性能之间的关系.
2 实验部分
2.1 催化剂的制备
介孔α-氧化锰的制备:将KMnO4(北京化工厂,分析纯)加入到顺丁烯二酸(国药集团化学试剂有限公司,分析纯)中配成溶液,静置24 h,抽滤、洗涤, 120°C干燥24 h,300°C焙烧2 h,即得α-MnO2.
xAu/α-MnO2(x%表示Au的投料负载量)的制备:将以上制备的α-MnO2加入到0.0025 mol·L-1HAuCl4·H2O(国药集团化学试剂有限公司,分析纯)溶液中,混合均匀,然后逐滴加入一定量1 mol·L-1的尿素(北京化工厂,分析纯)溶液,加热到75°C搅拌2 h,随后再加入0.5 mol·L-1NaBH4(天津市福晨化学试剂厂,分析纯)溶液搅拌1 h.过滤、洗涤、120°C干燥24 h,得xAu/α-MnO2.Au的实际负载量使用荷兰PANalytical公司生产的Magix(PW2403)型X射线荧光光谱仪分析,结果见表1.
2.2 催化剂表征
X射线粉末衍射(XRD)分析:使用Bruker-axs公司D8-ADVANCE射线衍射仪,入射光源为CuKα辐射,入射波长为0.15405 nm,管电压50 kV,管电流30 mA,扫描范围2θ=10°-80°,扫描速率3.5(°)· min-1;N2-吸附/脱附曲线用 Micromeritics公司的ASAP 2010测试仪在液氮温度下测得.测试前样品先于200°C,0.1 Pa条件下进行脱气处理.用BET法计算比表面积;用BJH法测定孔径分布.H2-程序升温还原(H2-TPR),高纯Ar(99.999%)作载气,5%(φ) H2作还原气,称取200 mg的样品,放入内径为6 mm的石英反应管内,载气流速为20 mL·min-1,升温速率为15°C·min-1,升温至950°C,保温30 min.还原气经过催化剂床层后,经过乙醇/液氮冷阱(-80°C)然后进入热导池检测.耗氢量通过CuO耗氢量的标准曲线校正.
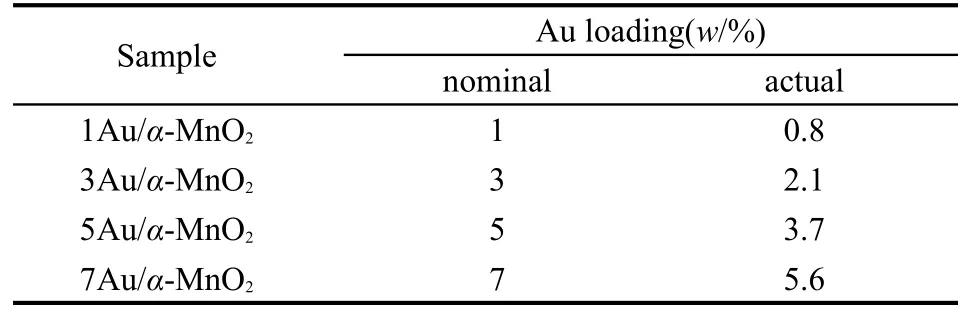
表1 xAu/α-MnO2样品的Au实际含量分析Table 1 Actual gold loadings of the xAu/α-MnO2catalysts
X射线光电子能谱(XPS)分析:使用VG Scientific公司的Esca-Lab-220i型X射线光电能谱仪,使用AlKα射线为激发源,电压20 kV,电流30 mA,基础真空度为3×10-7Pa,样品以污染碳的C 1s结合能(284.4 eV)作为内标校正样品的荷电效应.
2.3 催化剂的评价
CO的催化氧化活性是在常压固定床流动微型石英反应器中进行,催化剂添装量为200 mg(40-60目),并与200 mg(40-60目)石英砂混合,反应器内插入热电偶测量催化剂床层的温度,反应混合气组成为1.08%(φ)CO+空气,总流速为100 mL·min-1,空速30000 mL·(g-1·h-1).
以苯为代表VOCs的模型气体,将N2流过含苯的恒温鼓泡器,并与空气混合,获得2.0×10-3(φ)苯混合气浓度,空速120000 mL·(g-1·h-1).反应前后气体采用Shimadzu GC-8A气相色谱和天美GC-7900在线分析,其中O2、N2、CO和CO2等小分子气体采用TDX-01柱状分子筛导热检测器(TCD)分析,有机物采用SE-30柱子氢火焰离子化检测器(FID)分析. CO和苯的转化率通过反应前后CO和苯的含量变化计算.
3 结果和讨论
3.1 xAu/α-MnO2催化剂结构特点
图1为氧化锰载体及其负载不同Au含量样品xAu/α-MnO2(x=0-7,下同)的XRD谱图.结果表明,载体氧化锰(图1a)在2θ=29.0°,38.9°,49.7°,56.0°, 59.9°,69.08°处分别出现了对应四方晶型α-MnO2的(310)、(211)、(411)、(600)、(521)和(541)晶面的特征衍射峰(JCPDS 44-0141),其峰型较弥散,表明载体的结晶度比较低.加入Au后,xAu/α-MnO2样品中氧化锰载体的晶型结构不变,但其峰强随Au含量的增加略有增加,表明氧化锰的结晶度和颗粒略有增大,这可能与沉积-沉淀法制备负载Au过程中酸碱性变化有关.这点与Wu等14的结果相似,他们将贵金属负载在TiO2载体上,也发现载体的结晶度略有增加.

图1 不同Au负载量的xAu/α-MnO2的XRD图谱Fig.1 X-ray diffraction patterns of xAu/α-MnO2with differentAu loadings(a)α-MnO2;(b)1Au/α-MnO2;(c)3Au/α-MnO2;(d)5Au/α-MnO2; (e)7Au/α-MnO2
进一步研究表明,对于xAu/α-MnO2样品来说,当Au负载量较小(1%)(实际负载量为0.8%,见表1,下同)时,1Au/α-MnO2样品的XRD衍射图与载体氧化锰的峰强和峰型相似,未出现Au的特征峰,表明Au高度分散在氧化锰载体表面(<5 nm).当金负载量较大(3%-7%时,xAu/α-MnO2样品在2θ=38.2°, 44.4°,64.5°,78°处分别出现了对应Au0的(111)、(200)、(220)和(311)晶面的特征衍射峰,并且其衍射峰的强度随Au含量的增加而增加.通过Scherrer方程,依据Au0(200)晶面,计算出3Au/α-MnO2、5Au/ α-MnO2和7Au/α-MnO2样品中Au0的平均颗粒大小分别为6.5、13.8和16.5 nm.
3.2 xAu/α-MnO2催化剂织构特点
图2(A)为xAu/α-MnO2样品的N2-吸附/脱附等温曲线.结果表明,在p/p0<0.05低压处,载体α-MnO2具有明显的N2吸附/脱附,为典型的BDDT类I型等温线,表明样品存在微孔结构,此微孔可能主要来自于α-MnO2的隧道结构;在p/p0中间处,N2吸附缓慢上升;在p/p0>0.5高压处,N2具有明显的吸附,并形成滞后环,为典型的BDDT类IV型等温线,表明α-MnO2中存在介孔结构,其N2-吸附/脱附等温曲线属于H2类型,为多孔吸附质或均匀粒子堆积.负载Au后,xAu/α-MnO2样品的N2-吸附/脱附等温曲线与载体相似,表明样品的微孔和介孔结构不变,而且其吸附曲线的高度随Au含量变化不大,表明Au未阻塞α-MnO2载体的孔道,分布在α-MnO2孔道内外.图2(B)为xAu/α-MnO2样品的孔径分布图,xAu/ α-MnO2样品的最可几孔径分布为5.1-5.5 nm,加入Au对其最可几孔径分布影响不大,同样表明分布在α-MnO2载体上的Au未阻塞其孔径.
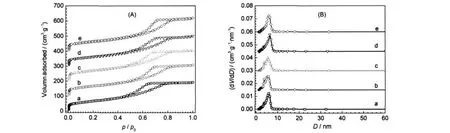
图2 不同Au负载量的xAu/α-MnO2的N2-吸附/脱附等温线(A)和孔径分布曲线(B)Fig.2 N2-adsorption/desorption isotherm(A)and pore size distribution(B)of xAu/α-MnO2with differentAu loadings(a)α-MnO2;(b)1Au/α-MnO2;(c)3Au/α-MnO2;(d)5Au/α-MnO2;(e)7Au/α-MnO2
表2是通过以上样品的N2-吸附/脱附等温线脱附曲线计算的孔结构参数,包括比表面积(SBET)、孔体积(VP)和平均孔径(dPD).结果表明,α-MnO2具有较高的比表面积,其比表面积高达248.3 m2·g-1,是普通方法制备的α-MnO2比表面积(23.8 m2·g-1)的10倍多.15随着Au含量的增加,xAu/α-MnO2样品的比表面积、孔体积和平均孔径变化不大,表明Au颗粒未阻塞载体α-MnO2的孔道,这与N2-吸附/脱附曲线(图2(A))和孔径分布曲线的结果一致(图2(B)).
为进一步测定不同xAu/α-MnO2中Au颗粒的大小,本研究通过TEM技术测定了xAu/α-MnO2表面Au颗粒大小,图3(a,b)分别为载体α-MnO2的TEM和高分辨TEM(HRTEM)图,图3(c,e,d,f)分别为3Au/α-MnO2和7Au/α-MnO2的TEM和HRTEM图.载体α-MnO2的TEM结果(图3a)表明,α-MnO2呈现出纳米棒型结构,其结晶度不高,这点与以上XRD结果(图1a)一致,HRTEM图(图3b)显示出现对应α-MnO2(200)晶面的晶格线,线宽为0.49 nm.从TEM结果可以看出载体α-MnO2本身并没有介孔,其N2-吸附/脱附曲线显示的介孔结构主要来自于α-MnO2纳米棒堆积而成,这点与Chen课题组16合成的α-MnO2结果一致.3Au/α-MnO2的TEM结果表明(图3c),3Au/α-MnO2的Au颗粒较小地分布在α-MnO2的表面,3Au/α-MnO2样品的HRTEM图(图3d)显示出对应α-MnO2(200)晶面的晶格线,晶格线宽为0.49 nm,还显示出对应Au(200)晶面的晶格线,线宽为0.20 nm,Au的粒径约3-8 nm.对于7Au/ α-MnO2来说,从图3(e,f)可以看出,7Au/α-MnO2的Au颗粒较大,粒径约在15-20 nm左右,分布不均匀,有的颗粒略大于XRD测定的结果,这可能是由于Au颗粒聚集.
3.3 xAu/α-MnO2催化剂的还原性能
为了研究氧化锰载体及其负载金样品的还原性能,对其进行了H2-TPR测定.图4为氧化锰载体及其负载金xAu/α-MnO2样品的H2-TPR谱图.α-MnO2载体(图4a)在360和500°C分别出现了一个宽的和一个弱的还原峰,根据文献,17分别对应MnO2→Mn3O4和Mn3O4→MnO的还原.由于将MnO还原成金属态Mn0需要较大的潜能,所以到850°C还原结束时,没有MnO→Mn0的还原峰,即,MnO为MnO2还原的终态,这点我们通过还原后的XRD证实了这点(没有显示).对于α-MnO2载体,通过耗氢量计算表明其H/Mn的摩尔比例,约为1.58,低于MnO2完全还原成MnO(MnO2+H2→MnO+H2O)的理论还原H/Mn比例(2.0).表明在α-MnO2中存在混合价态的Mn离子(即,Mn3+和Mn4+),这与以下的XPS结果一致.
从图中可以看出,与载体α-MnO2的还原过程相比,xAu/α-MnO2样品的还原峰明显向低温方向移动,而且只出现一个还原峰,峰面积有所增大(图4 (b,e));即加入Au后,xAu/α-MnO2样品的还原性、还原速度和还原程度在增加.xAu/α-MnO2样品在320-340°C处出现的还原峰,应属于MnO2→MnO一步还原.17当然,少量的Au在低于400°C也会被还原,它们的还原峰被α-MnO2的还原峰所覆盖.负载Au使α-MnO2样品的还原温度明显降低,促进了xAu/α-MnO2样品的还原,这可能是由于贵金属(如Pt、Au、Rh和Ag)通过对H2的溢流作用和促进α-MnO2样品中晶格氧的移动性、活化和提高了MnO2的还原性,18另一方面,也表明在Au和α-MnO2之间存在强的相互作用,这点与以下XPS结果一致,Li及其合作者19在研究Ag/MnO2催化剂时也得出相同的结论.在xAu/α-MnO2样品中,3Au/α-MnO2样品低温还原温度最低,也就是说,其具有最高的氧化还原能力.
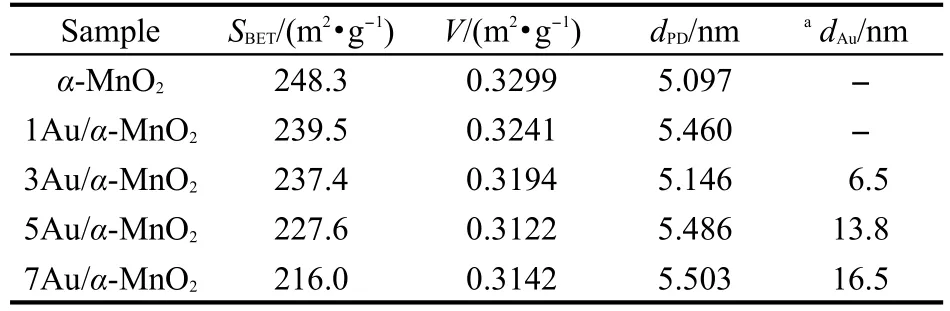
表2 xAu/α-MnO2样品的比表面积、孔体积、平均孔径及Au颗粒大小Table 2 Surface areas,pore volume,pore diameter and meanAu particle sizes of the xAu/α-MnO2catalysts

图3 xAu/α-MnO2样品的TEM和高分辨TEM(HRTEM)图Fig.3 TEM and high-resolution TEM(HRTEM)images of xAu/α-MnO2 (a,b)α-MnO2;(c,d)3Au/α-MnO2;(e,f)7Au/α-MnO2
3.4 xAu/α-MnO2催化剂表面离子价态
图5为xAu/α-MnO2样品O 1s、Au 4f和Mn 2p的XPS谱图.表3为各个XPS峰所对应的峰位置和不同价态物种的表面含量比值.图5(A)表明在Eb≈530-533 eV处存在一个O 1s不对称峰.通过解峰,此峰可以分解成Eb≈530-533 eV两个峰,分别对应两个不同的氧物种Oα和Oβ.Oα结合能位于529-530 eV,为表面晶格氧物种(O2-),Oβ结合能位于531-533 eV,为表面吸附氧物种或O-),20其主要来源于xAu/α-MnO2样品表面氧空位对O2的吸附.21Au的负载明显影响xAu/α-MnO2中Oα和Oβ的浓度(图5(A)和表3),随着Au含量从0.8%(1Au/α-MnO2)增加到5.6%(7Au/α-MnO2),Oα/Oβ摩尔比从1.22逐渐也逐渐增加到5.26.也就是说,随Au含量增加, xAu/α-MnO2中晶格氧的量(Oα)逐渐升高,而缺陷氧/吸附氧的量(Oβ)逐渐降低.这表明,与载体α-MnO2相比,加入Au可以明显提高载体α-MnO2中晶格氧物种的浓度,即增加Mn4+的浓度,这与以下Mn 2p的XPS结果一致.

图4 不同金负载量xAu/MnO2的TPR图Fig.4 H2-TPR patterns of of xAu/α-MnO2with differentAu loadings(a)α-MnO2;(b)1Au/α-MnO2;(c)3Au/α-MnO2;(d)5Au/α-MnO2; (e)7Au/α-MnO2

图5 xAu/α-MnO2样品中O 1s(A),Mn 2p(B),Au 4f(C)的XPS图谱Fig.5 O 1s(A),Mn 2p(B),andAu 4f(C)XPS spectra of xAu/α-MnO2 (a)α-MnO2,(b)1Au/α-MnO2,(c)3Au/α-MnO2,(d)5Au/α-MnO2,(e)7Au/α-MnO2
图5(B)为xAu/α-MnO2样品中Mn 2p的XPS谱图.所有样品的Mn 2p3/2和Mn 2p1/2峰分别位于Eb≈641,642,652.7和653.7 eV.Mn 2p的自旋轨道分裂能约为11.7 eV,这与MnO2和Mn2O3相似.22根据文献Eb≈641 eV(Mn 2p3/2)和642(Mn 2p3/2)处的结合能分别属于Mn3+和Mn4+物种.23即,xAu/α-MnO2中的Mn主要以+3价和+4价存在,这与TPR结果一致.但是前面XRD(图1)没有检测到Mn2O3(Mn3+)的存在,这可能与XPS技术与XRD技术在检测化学相上的灵敏性有关.根据XPS技术测定的表面Mn4+/Mn3+比例结果示于表3.结果表明,随着Au含量从0.8% (1Au/α-MnO2)增加到5.6%(7Au/α-MnO2),xAu/α-MnO2样品表面Mn4+/Mn3+的原子比例从1.54增加到3.18.即随着Au负载量的增加,xAu/α-MnO2样品中Mn的氧化态在增加,表明MnO2与Au之间存在强的相互作用.基于电中性原理,可以推测在xAu/ α-MnO2样品中存在氧空位,并且随Au含量的增加而降低,这与以上O 1s的XPS结果一致(图5(A)).
图5(C)为xAu/α-MnO2样品中Au 4f的XPS谱图.xAu/α-MnO2样品中Au 4f中存在两个明显不对称的峰,分别位于Eb≈84.0,88.0 eV,它们可以分解成:Eb≈83.7,85.2 eV和Eb≈87.5,89.0 eV,前两峰是Au 4f7/2的特征峰,后两峰为Au 4f5/2的特征峰.Eb≈83.7,89.0 eV属于Au0,而Eb≈85.2,89.0 eV属于Au3+.22即在xAu/α-MnO2样品上同时存在Au0和Au3+,并且随着Au含量从0.8%(1Au/α-MnO2)增加到5.6%(7Au/α-MnO2),xAu/α-MnO2样品中的Au3+/ Au0比例从0.22增加到0.46(表3).分析其比例,可以看出,Au含量较低时,Au主要以金属态Au0分散在α-MnO2载体表面,Au含量较高时,Au以金属态Au0和氧化态Au2O3(Au3+)分散在α-MnO2载体表面.但是人们研究表明,当焙烧温度大于300°C时,氧化态Au3+容易分解成金属态Au0,23而本研究中,xAu/ α-MnO2样品的制备是在空气氛围下400°C焙烧了2 h,高于氧化金分解温度,而xAu/α-MnO2样品催化剂上存在氧化态的Au3+,这可能是MnO2与Au之间存在强的相互作用(以上TPR和XPS结果),MnO2中部分氧物种转移到Au上将其氧化成氧化态Au.
3.5 xAu/α-MnO2催化剂催化氧化性能
图6为xAu/α-MnO2(x=0-7)催化剂上催化氧化CO的活性.结果表明,低于70°C时,xAu/α-MnO2催化剂催化活性较低,当温度高于70°C时,催化活性随温度明显升高.α-MnO2载体的催化活性明显低于Au负载型样品xAu/α-MnO2(x≠0)的催化活性,表明加入Au能明显提高催化剂的催化活性.随着Au浓度从0增加到3%,xAu/a-MnO2催化剂的活性明显增加,当Au浓度进一步增加,其催化活性明显下降,即3Au/α-MnO2具有最高的催化活性.

表3 xAu/α-MnO2样品的表面原子结合能(Eb)及Mn4+/Mn3+、Au3+/Au0和Oα/Oβ的摩尔比Table 3 Binding energies(Eb)of surface elements and molar ratios of Mn4+/Mn3+,Au3+/Au0,and Oα/Oβover the xAu/α-MnO2catalysts
对于VOCs气体来说,芳香族类化合物是最难催化氧化的物种,本研究选择此类化合物中的苯代表VOCs气体,研究其在xAu/α-MnO2催化剂上催化氧化的活性,结果如图7所示,产物分析及碳平衡计算表明,CO2和H2O是催化氧化反应唯一的产物.结果表明,α-MnO2载体的催化活性明显低于xAu/ α-MnO2(x¹0)催化剂.表明Au负载在α-MnO2催化剂上对催化氧化苯具有重要的作用.在xAu/α-MnO2催化剂上,催化活性随Au负载量从0.8%(x=1)增加到2.1%(x=3)而增加,当Au含量从2.1%(x=3)进一步增至5.6%(x=7)时,其催化活性减小,即,3Au/ α-MnO2具有最佳催化氧化苯活性.以上结果表明, xAu/α-MnO2催化剂随Au负载量变化催化氧化CO和苯的活性规律相似,其活性顺序如下:
3Au/α-MnO2>1Au/α-MnO2>5Au/α-MnO2>7Au/
α-MnO2>α-MnO2

图6xAu/α-MnO2催化剂上催化氧化CO的转化率随温度的变化Fig.6 Carbon monoxide conversion as a function of temperature over the xAu/α-MnO2catalysts SV=60000 mL·(g-1·h-1)
即适当的Au负载可以获得较高的催化氧化CO和苯活性,3Au/α-MnO2的活性最高,其CO和苯的T90(催化氧化CO和苯转化率90%时温度)分别为80°C和200°C.3Au/α-MnO2的高催化活性,可能与以下原因有关.首先,大多研究表明,Au颗粒大小是决定Au负载型催化剂催化氧化CO活性的关键因素.Hvolbaek等24研究认为,高度分散的Au具有低配位数,Au与载体接触的界面是CO催化氧化的活性位,反应活性位数目直接与载体和Au接触的li(li边界周长)成正比,即与di2(di为颗粒直径)成反比,因此,降低Au颗粒大小可以极大地增加Au的配位数和活性位数目,颗粒越小,活性位越多,活性越高.同样,对于苯的催化氧化来说,高度分散的Au颗粒有利于活化O2和苯,Ahn等25研究氧化物(Al2O3、Fe2O3和Co3O4)负载Au催化剂认为高度分散Au催化剂有利于O2的分解,从而促进“氧化-还原循环”反应的进行.Lambert等26也认为负载型(Pd、Ag和Cu)催化剂中,活性组分的分散度是决定苯催化氧化活性的重要条件.以上XRD研究表明,3Au/ α-MnO2催化剂上Au颗粒大小约为6.5 nm,而其它含量xAu/α-MnO2(x>3)样品中,Au颗粒大小为13.8-16.5 nm,明显大于3Au/α-MnO2催化剂,所以3Au/α-MnO2催化剂活性明显优于其它xAu/α-MnO2催化剂,虽然1Au/α-MnO2催化剂上Au颗粒也较小(<5 nm),但其活性较3Au/α-MnO2低,这可能与其Au含量较低有关.Haruta等13认为较小的Au颗粒和适当Au负载量是获得高催化氧化活性的前提.
其次,xAu/α-MnO2催化剂中负载不同价态的Au也可能是它们催化活性有差别的原因.目前人们对于Au催化剂上CO催化氧化机理争论的焦点主要集中在反应活性中心(金属态Au0或氧化态Auδ+).27Goodman及其合作者28使用扫描隧道显微镜(STM)和光谱结合研究,认为CO催化氧化反应活性应归因于小粒径金的量子效应.Henao等29通过红外光谱分析,在2090 cm-1处观察到了CO与Au0结合的红外振动光谱,他们认为金属态Au0为CO的吸附位,催化活性较高的催化剂通常都含有金属态Au0,而氧化态Auδ+并不能形成稳定的活性位.Haruta及其合作者13认为,金属态Au0比金属态Au0和氧化态Auδ+的混合物种具有更高的活性.与CO催化氧化相似,贵金属存在的价态对催化剂催化氧化活性影响较大,Taralunga等30认为,对于负载型贵金属催化剂,还原态贵金属比氧化态对催化氧化苯系物更具活性.前面XPS结果表明(图5(C)和表3),随着Au含量的增加,xAu/α-MnO2中氧化态Au3+在增加,而还原态的Au0在减小,而3Au/α-MnO2主要以还原态Au0存在,这可能也是其活性较高的原因之一.
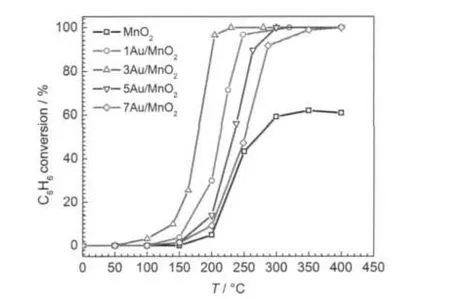
图7 xAu/α-MnO2催化剂上催化氧化苯的转化率随温度的变化Fig.7 Benzene conversion as a function of temperature over the xAu/α-MnO2catalysts SV=120000 mL·(g-1·h-1)
另外,对于具有氧化-还原性载体负载Au催化剂来说,除了Au颗粒大小和价态对其催化活性影响较大外,载体的作用也很重要.如Grisel等31将Au负载在此类载体上,虽然Au颗粒大小相似,但是由于载体活化反应物的能力不同,Au负载型催化剂催化氧化CO活性差别较大.通常,CO和碳氢化合物在具有氧化还原性载体负载Au催化剂上的催化氧化反应依Mars-van Krevelen机理进行,32其详细的催化氧化机理虽然不是完全清楚,通常认为对于CO催化氧化反应途径可能是:CO在Au与载体界面被活化,形成吸附态的CO*,O2直接吸附于氧化物载体表面氧空位上,O-O键断裂,形成吸附态的氧原子O*,O*与CO*在催化剂表面反应生成CO2,即Au主要活化CO,而氧化物载体活化O2,为供氧载体.同样,对于苯的催化氧化反应,大多研究认为,30反应不仅发生在Au的表面,而且发生在Au和MnO2的界面,载体α-MnO2同样是供氧体,而贵金属Au主要是吸附活化苯.所以载体活化O2的能力与其催化氧化活性明显相关,而载体活化O2的能力与其表面氧空位和氧化还原性能密切相关.通常氧化还原性和表面氧空位浓度越高,其活化O2能力也越强.33众所周知,氧化锰具有深度和快速的氧气“贮存/释放”能力,在反应气氛中,锰可以形成不同的氧化态,如:Mn2+/Mn3+或Mn3+/Mn4+,通过与反应气氛中还原和氧化物种的作用,其具有较好的“氧化/还原性能”和“氧移动性”.我们认为在xAu/α-MnO2样品中存在Au和载体之间强的相互作用,导致其氧的移动性提高,从而获得高的还原性能和催化活性.相似的结果在Au/CeO2和Au/CuO催化系统中有报道.34前面XPS结果表明,随着Au浓度的增加,xAu/α-MnO2催化剂Mn4+浓度和表面晶格氧(O2-)在增加,而表面吸附氧物种,即空位氧或O-)浓度在减少,即对于xAu/α-MnO2样品来说,具有适当Au负载量的xAu/ α-MnO2样品中存在大量的Mn3+和空位氧(O2-2或O-),即存在“O→Mn”电子迁移,形成具有反应活性的亲电子空位氧物种或O-),这点与Arena等35研究MnCeOx样品中电子迁移的现象结果相似.从 xAu/α-MnO2氧化还原性结果(H2-TPR)看,适量Au负载量明显提高α-MnO2的氧化还原性能,其中3Au/ α-MnO2样品具有最强的氧化还原性能.即,Au和α-MnO2之间存在强的相互作用.综合3Au/α-MnO2样品表征结果可以看出,3Au/α-MnO2催化剂具有合适的活性氧物种和最强的氧化还原性能,使其具有较强的活化氧能力,从而有利于催化反应的“氧化-还原的循环”进行,具有较强的催化氧化活性.另外,此样品具有较小的Au颗粒和存在大量的还原态Au0,以上因素使之具有最高的催化氧化CO和苯活性.
4 结论
采用沉积-沉淀法制备了α-MnO2负载Au催化剂xAu/α-MnO2(x=0-7).并且评价了其催化氧化CO和苯的活性.
(1)XRD结果表明,当Au负载量较小时(x≠3 (2.1%)),xAu/MnO2样品中的Au高度分散在氧化锰载体表面(<6.5 nm),当Au负载量较大(x>3(2.1%))时,xAu/MnO2样品中Au颗粒大小随Au含量增加而明显增大.N2-吸附/脱附结果表明,Au负载量对xAu/ α-MnO2样品的比表面积、孔溶和孔径影响不大,表明Au均匀分布在α-MnO2载体表面.
(2)XPS结果表明,随着Au负载量的增加,xAu/ α-MnO2中的Oα/Oβ、Mn4+/Mn3+和Au3+/Au0的摩尔比在增加,表明其晶格氧、Mn4+和Au3+的浓度随Au含量增加而增加.由于贵金属的溢氢作用,Au明显促进xAu/α-MnO2氧化还原能力,其中3Au/α-MnO2具有最高的氧化还原性.
(3)负载Au明显影响xAu/α-MnO2的催化活性, xAu/α-MnO2的催化性能与Au的分散性、低温氧化还原性能及表面氧物种的种类密切相关,其中3Au/ α-MnO2显示出最佳活性,其催化氧化CO的T90= 80°C,苯的T90=200°C.
(1) Gardner,S.D.;Hoflund,G.B.;Schryer,D.R.;Schryer,J.; Upchurch,B.T.;Kielin,E.J.Langmuir 1991,7,2135.
(2) Li,Q.X.;Zhou,X.J.;Li,J.G.;Xu,C.J.Acta Phys.-Chim.Sin. 2010,26,1488.[李巧霞,周小金,李金光,徐群杰.物理化学学报,2010,26,1488.]
(3) Liu,Y.J.;Zhang,J.J.;Li,N.;Lin,B.X.Acta Phys.-Chim.Sin. 1999,15,97.[刘英骏,张继军,李 能,林炳雄.物理化学学报,1999,15,97.]
(4) Spivey,J.J.Ind.Eng.Chem.Res.1987,26,2165.
(5) Zwinkels,M.F.M.;Jaras,S.G.;Menon,P.G.;Griffin,T.A. Cat.Rev.-Sci.Eng.1993,35,319.
(6)Taylor,S.H.;Heneghan,C.S.;Hutchings,G.J.;Hudson,I.D. Catal.Today 2000,59,249.
(7) Kulshreshtha,S.K.;Gadgil,M.M.Appl.Catal.B 1997,11,291.
(8) Luo,M.F.;Yuan,X.X.;Zheng,X.M.Appl.Catal.A 1998,175, 121.
(9) Ye,Q.;Zhao,J.S.;Huo,F.F.;Wang,J.;Cheng,S.Y.;Kang,T. F.;Dai,H.X.Catal.Today 2011,175,603.
(10)Haruta,M.;Kobayashi,T.;Sano,H.;Yamada,N.Chem.Lett. 1987,16,405.
(11) Zhang,X.;Shi,H.;Xu,B.Q.Catal.Today 2007,122,330.
(12) Shao,J.J.;Zhang,P.;Song,W.;Huang,X.M.;Xu,Y.D.;Shen, W.J.Acta Chim.Sin.2007,65(18),2007. [邵建军,张 平,宋 巍,黄秀敏,徐奕德,申文杰.化学学报,2007,65(18), 2007.]
(13) Haruta,M.;Tsubota,S.;Kobayashi,T.;Kageyama,H.;Genet, M.J.;Delmon,B.J.Catal.1993,144,175.
(14)Wu,Z.B.;Sheng,Z.Y.;Liu,Y.;Wang,H.Q.;Mo,J.S. J.Hazard.Mater.2011,185,1053.
(15) Kijima,N.;Yasuda,H.;Sato,T.;Yoshimura,Y.J.Solid State Chem.2001,59,94
(16) Chen,Y.;Liu,C.;Li,F.;Cheng,H.M.J.Alloy.Compd.2005, 397,282.
(17) Carno,J.;Ferrandon,M.;Bjornbom,E.;Jaras,S.Appl.Catal.A 1997,155,265.
(18) Tsuji,Y.;Imamura,S.In New Aspects of Spillover Effect in Catalysis;Inui,T.;Fujimoto,K.;Uchijima,T.;Masai,M.Eds. Elsevier:Amsterdam,1993;p 405.
(19)Xu,R.;Wang,X.;Wang,D.S.;Zhou,K.B.;Li,Y.D.J.Catal. 2006,237,426.
(20) Hamoudi,S.;Larachi,F.;Adnot,A.;Sayari,A.J.Catal.1999, 185,333.
(21) Madier,Y.;Descorme,C.;Le Govic,A.M.;Duprez,D.J.Phys. Chem.B 1999,103,10999.
(22) Muilenbergy,G.E.Handbook of X-Ray Photoelectron Spectroscopy;Perkin-Elmer Corporation:Minnesota,1979.
(23) Zhen,M.;Steve,H.O.;Sheng,D.J.Mol.Catal.A-Chem. 2007,273,186.
(24) Hvolbaek,B.;Janssens,T.V.W.;Clausen,B.S.;Falsig,H.; Christensen,C.H.;Norskov,J.K.Nanotoday 2007,2,14.
(25)Ahn,H.G.;Lee,D.J.Res.Chem.Intermed.2002,28,451.
(26) Lambert,S.;Cellier,C.;Gaigneaux,E.M.;Pirard,J.P.; Heinrichs,B.Catal.Commun.2007,8,1244.
(27) Finch,R.M.;Hodge,N.A.;Hutchings,G.J.;Meagher,A.; Pankhurst,Q.A.;Siddiqui,M.R.H.;Wagner,F.E.;Whyman, R.Phys.Chem.Chem.Phys.1999,1,485.
(28)Valden,M.;Lai,X.;Goodman,D.W.Science 1998,281,1647.
(29) Henao,J.D.;Caputo,T.;Yang,J.H.;Kung,M.;Kung,H.H. J.Phys.Chem.B 2006,110,8689.
(30) Taralunga,M.;Mijoin,J.;Magnoux,P.Appl.Catal.B 2005,60, 163.
(31) Grisel,R.J.H.;Nieuwenhuys,B.E.J.Catal.2001,199,48.
(32) Mars,P.;van Krevelen,D.W.Chem.Eng.Sci.Spec.Suppl. 1954,3,41.
(33) Liu,H.;Kozlov,A.I.;Kozlova,A.P.;Shida,T.;Iwasawa,Y. Phys.Chem.Chem.Phys.1999,1,2851.
(34) Venezia,A.M.;Pantaleo,G.;Longo,A.;Carlo,G.D.;Casaletto, M.P.;Liotta,F.L.;Deganello,G.J.Phys.Chem.B 2005,109, 2821.
(35) Arena,F.;Trunfio,G.;Negro,J.;Fazio,B.;Spadaro,L.Chem. Mater.2007,19,2269.
June 13,2011;Revised:September 13,2011;Published on Web:September 29,2011.
Highly Active Au/α-MnO2Catalysts for the Low-Temperature Oxidation of Carbon Monoxide and Benzene
YE Qing*HUO Fei-Fei YAN Li-Na WANG Juan CHENG Shui-Yuan KANG Tian-Fang
(College of Environmental and Energy Engineering,Beijing University of Technology,Beijing 100124,P.P.China)
α-MnO2-supported gold catalysts(xAu/α-MnO2,x=0-7(corresponding to the Au loading(mass fraction)of 0-7%)were prepared by a deposition-precipitation method using urea as a precipitation agent and characterized by different techniques such as X-ray diffraction(XRD),N2adsorption-desorption measurements,transmission electron microscopy(TEM),X-ray photoelectron spectroscopy(XPS),and H2temperature-programmed reduction(TPR).The catalytic activities of the materials were evaluated for the oxidation of CO and benzene.The Au particle size was found to be related to the Au loading of the xAu/ α-MnO2samples and increased with Au loading.XPS results showed that the mole ratios of O2-/or O-), Mn4+/Mn3+and Au3+/Au0increased upon the addition of Au.The loading of gold over α-MnO2significantly modified the catalytic activities.The catalytic performance of xAu/α-MnO2strongly depended on the Au loading,and 3Au/α-MnO2gained the best activity at T90=80°C and T90=20°C for the catalytic oxidation of CO and benzene,respectively.The excellent performance of 3Au/α-MnO2is associated with highly dispersedAu, good low-temperature reducibility,andasynergism at the interfacebetween theAu andMnO2nanodomains.
α-MnO2supported gold catalyst;Low-temperature reducibility;Synergistic action; Carbon monoxide oxidation;Benzene combustion
10.3866/PKU.WHXB20112872
*Corresponding author.Email:yeqing@bjut.edu.cn;Tel:+86-10-67391659.
The project was supported by the National Natural Science Foundation of China(20777005),Natural Science Foundation of Beijing,China (8082008)and Beijing Municipal Foundation for Excellent Person ofAbility,China(20071D0501500210).
国家自然科学基金(20777005),北京市自然科学基金(8082008)和北京市组织部优秀人才基金(20071D0501500210)资助项目
O643
猜你喜欢
化学工程师(2023年1期)2023-02-17
理化检验-化学分册(2020年12期)2021-01-26
云南化工(2020年11期)2021-01-14
矿产综合利用(2020年1期)2020-07-24
上海农业科技(2019年1期)2019-02-22
中国果业信息(2018年5期)2018-01-17
中学化学(2017年4期)2017-07-07
新课程·中旬(2016年12期)2017-05-08
试题与研究·教学论坛(2017年8期)2017-03-23
河北科技大学学报(2015年5期)2015-03-11
