
微囊藻毒素检测方法研究进展*
2012-03-20 00:40崔萌萌马康何雅娟杨兰
化学分析计量 2012年6期
崔萌萌,马康,何雅娟,杨兰
(1.中国计量科学研究院,化学计量与分析科学研究所,北京 100013; 2.北京化工大学化工资源有效利用国家重点实验室,北京 100029)
蓝藻是现存研究发现的毒性最高、污染范围最大的淡水藻类之一。水体中的蓝藻在非常短的时间内大量生长并聚集就会产生一种异常生态现象,称为水华。藻类生长繁殖速度会在水体富营养化的情况下异常加快,水华在这种情况下就会爆发,这已经成为了一个全球性的问题[1]。有关组织通过数据分析发现,亚州地区和欧洲地区湖泊富营养化已经超过了50%,北美洲和南美洲略微低点,分别是48%和41%,我国则高达67%[2]。近年来,我国几大淡水湖泊都发生过蓝藻水华大量爆发的情况。虽然藻类植物释放到天然水体中的藻毒素含量非常低,但由于藻类水华非常严重而且范围较广,使水体中的藻毒素的含量呈现上升的趋势,从而危及人们的饮水安全和健康。蓝藻中最典型的毒素就是微囊藻毒素(MC),它是一类肝毒素,它们的结构是由7个氨基酸组成的环状多肽[3],结构通式:环-(D-丙氨酸-L-X-D-赤-甲基-β-D-异天冬氨酸-L-Z-Adda-D-异谷氨酸-N-甲基脱氢丙氨酸),包括3个右旋(D-)氨基酸、两个可以改变的左旋(L-)氨基酸,其中N-甲基脱氢丙氨酸是一种含有α、β不饱和双键的特殊的氨基酸;Adda结构为3-氨基-9-甲氧基-2,6,8-三基酸-10-苯基-4(E),6(E)-二烯酸。
在MC中表达生理活性的必需基团是Adda结构,由于Adda部分含有共轭双键,MC在某些条件下会转化为[4(E),6(E)-Adda]MC和[4(Z),6(E)-Adda]MC两种空间异构体,[4(Z),6(Z)-Adda]MC还未见报道。研究者至今已发现了80多种MC的同分异构体,其中存在普遍、毒性较强的是MC–LR,MC–RR,MC–YR,(L,R,Y—亮氨酸、精氨酸和酪氨酸)[4–5],对它们的研究也较多。MC–LF和MC–LW分别含有苯丙氨酸和色氨酸。在MC同分异构体的分子结构中,Adda侧链是MC中毒性表达的重要基团,失去Adda侧链后MCs的毒性会降低[6]。MC中常见的具有代表性的异构体分子结构如图1所示。
对比图1中MC同分异构体的结构可以看出虽然它们的结构大体相同,但却存在着明显的区别,因而物理性质和化学性质都会存在较大的区别,分离时难度较大。
MC是具有非常强烈毒性的肝毒素,同样具有促癌作用,很少的量就会对人和生物体的安全构成威胁。其化学性质非常稳定,在强酸、强碱和高温条件下都不易分解,而且其具有强致毒性,又因其分布较广,故其危害性不容忽视。医学研究显示,我国江苏和广西很多地区的居民由于长期饮用含微量MC的饮用水而导致肝癌发病率较高。
人们对MC–LR和总MC限量分别为0.32 μg/L和0.88 μg/L(以成年人为标准)。因为MC–LR在MC中的毒性是最大的,而且促肿瘤作用也非常明显,其它的藻毒素可能不具有较强的代表性,所以目前人们大多都以MC–LR作为代表用于饮用水中藻毒素含量的限制。世界卫生组织认定饮用水中藻毒素的标准限值为1.0 μg/L(以MC–LR为代表),国家环境保护总局近期颁布的《中华人民共和国国家标准》(GB 3838–2002)中就指出:“居民集中式生活饮用水和地表水源地中MC以MC–LR代表的限值为1.0 μg/L”。
对MC的分离和检测方法的研究之前已经有许多报道。目前普遍采用的方法可以将其归纳为化学分析法、生物法和生物化学法[7]等。
1 化学分析检测法
化学分析检测法主要有高效液相色谱(HPLC)、高效液相色谱串联质谱(HPLC–MS)、薄层层析色谱(TLC)[8]、气相色谱[9]串联质谱(GC–MS)联用、毛细管电泳(CE)[10]等方法。
1.1 高效液相色谱法
在化学检测方法中目前使用最为普遍是HPLC法[11–12]。目前,WHO、美欧等工业发达国家和地区以及我国权威机构多数推荐使用HPLC法对MC进行检测分析。该方法具有重现性好、准确度和灵敏度高的优点,且能同时对不同的MC同分异构体进行分析。目前研究报告中有很多关于HPLC检测MC都集中在对样品的前处理、色谱分析条件、淋洗剂、洗脱液、SPE柱、浓缩定容过程等的优化处理上。
分离MC的固定相有很多种,其中有报道的包括反相C18柱、离子交换柱、酰胺C16柱、内部界面反相柱、XAD–2等大孔吸附树脂[13]等,研究中大多选用反相C18柱,此种色谱柱的优点是其固定相稳定、应用广泛、可在多种溶剂中使用,一般的C18柱pH范围通常保持在2~8。在检测MC的实验中,流动相多采用甲酸或三氟乙酸酸化的水溶液和乙腈或甲醇的梯度洗脱,这可以使大多数MC分离。Esme[14]等在用HPLC检测MC时就只采用甲醇作为流动相的有机相,并对MC的一系列异构体进行了分离。房祥军[15]等在检测MC时发现在238 nm下甲醇的本体吸收值高于乙腈。但采用乙腈做流动相时,分离藻毒素柱压小,基线稳定,峰形尖锐,分离效果明显优于甲醇–水流动相。当流动相中加入一定量的三氟乙酸后,峰形较好,分离度提高。而且酸度越低,对色谱柱的保护越有利。通常高效液相色谱仪都连接有不同检测器,其中使用最多的是紫外(UV)和光电二极管阵列检测器(DAD)用紫外检测器进行高效液相色谱分析,能为定量定性提供有效的数据,成为鉴定MC的一种可行的方法。Christine等[16]用DAD检测器对MC进行纯度检测时,证实比用其它检测器进行检测得到的结果更精确。由于MC上特殊氨基酸Adda上的烯烃作为主要的发色团,通常在238 nm处MC有最大吸收的特征光谱,在222 nm处产生最大吸收的色氨酸[17]。闫海等[18]在使用二级管紫外检测器的液相色谱上进行测定,发现MC–LR和MC–RR在238 nm波长下吸收峰均最大。
MC在HPLC条件下的检出限为1 mg/L,而MC在天然水体中含量仅为μg/L水平,故水样一般都要进行前处理,具体方法是通过富集柱进行浓缩洗脱后用HPLC进行测定。Lee[19]等用高效液相色谱方法检测水样中的MC–LR,MC–RR及MC–YR时,发明了一种可以改变柱效的新方法,在不需要对水样进行预净化的情况下,将过滤后的水样直接在Zorbax CN预置柱进行在线浓缩,浓缩后的被分析物经过反相洗脱,在Luna C18柱上分离,这种方法的分析速度、准确度和精密度都非常好,通过将被测毒素的峰面积与标准毒素的峰面积进行比较可对MC定量。
HPLC分析检测MC的缺点:需要大型贵重仪器,且检测过程复杂耗时,检测时需要MC标准物质,目前已发现的80多种MC同分异构体大多数缺乏标准物质,MC同系物的定量只能参照MC–LR。由于各实验室的检测程序和条件的差异性,可能对MC分析测定对比分析产生一定的影响。
1.2 液相色谱–质谱联用技术
高效液相色谱–质谱法简便、灵敏,广泛用于监测含有MC的环境样品。液相色谱–质谱技术具有良好的选择性,该方法对含有多种藻类毒素的复杂样品进行分离和鉴定时可以使用。目前只有很少的藻毒素有相应的毒素标准品,但当被测毒素分子量已知时,就可通过准确检测出检测物的结构信息对其进行定性分析,对毒素定量分析时可以达到更高的精度和准确度[20]。此法的优点是在质谱检测器上测定分子量和洗脱化合物,其检测结果具有较好的特异性,不足之处是实验成本较高。之前在只用光电二极管阵列检测器(PDA)下不能分辨得到不同多肽类藻毒素的情况下,现在运用质谱检测器都可以区分开来,其结果如下:MC–LR,MC–YR,MC–RR的检测下限分别为37,42,23 ng/L[21]。
虞锐鹏[22]等用水–甲酸–乙腈作为流动相,采用液相色谱–电喷雾质谱(HPLC–MS–ESI)法对水中的MC进行测定,此方法的检出限达到0.01 μg/L,线性定量范围为0.02~20.0 μg/L。此种实验的研究可为水质检测领域提供快速、灵敏和准确的分析方法。Werawan[23]等利用液相色谱–电喷雾质谱检测了MC–LR的含量。采用氨水–乙腈溶液在pH 9.7的条件下进行线性洗脱,方法检出限达到0.1 μg/mL。
张昱等[24]通过固相萃取法对MC进行提取和富集,然后使用HPLC/ESI–MS测定水中的MC–RR,MC–YR和MC–LR,回收率接近90%,线性范围达到3个数量级以上(0.5~500 ng/L)。王超[25]等建立了一种液相色谱–二极管阵列检测器(LC–DAD)/离子阱质谱(ITMS)对水中5种MC的分析方法。水中的MC经固相萃取富集和纯化,经液相色谱分离后,采用DAD和ITMS定性分析和AD定量分析。水中5种MC的检出限为0.1 μg/L,3个质量浓度加标水平(0.2,0.8,5 μg/L)的平均回收率为(52.2%~115.2%),相对标准偏差为1.2%~10.0%。该方法在进行定性定量分析时可以多种角度同时进行,并可对水中不同的MC进行检测。超高效液相色谱串联质谱(UPLC–MS/MS)技术是近几年来才发展起来的,此方法对分析速度的提高有很大的改善。王静等[26]在检测水体中MC–LR,MC–RR,MC–LW,MC–LF时,样品经SPE小柱后,UPLC–MS/MS进行检测,4种MCs的分离与检测在5 min之内就可完成。Ortelli等[27]运用超高液相色谱–飞行时间质谱(UPLC–Q–TOF)测定了MC的含量,该方法简便、快速、稳定,样品经超声波提取、过滤后无需任何纯化步骤,直接进仪器分析,而且检测的灵敏度高,水中的MC检出限为0.1 μg/L,微藻样品中的MC检出限达0.1~0.2 μg/g。茅海琼等[28]发展了超高效液相色谱–电喷雾串联四极杆质谱快速测定水中MC–LR的方法。水样经过前处理后,应用超高效液相色谱–电喷雾串联四极杆质谱仪多离子反应监测(MRM)对MC–LR进行定量检测,该方法灵敏度较高,快速简单易于操作,有利于进行宽浓度范围准确定量。张明[29]等研究固相萃取–超高效液相色谱–电喷雾串联三重四极杆质谱联用技术分析水中9种MC的方法。样品经SPE提取和净化后,以UPLCTMBEH C18色谱柱为分离柱,以0.1%甲酸乙腈溶液作为有机流动相和0.1%甲酸水溶液作为水相进行梯度洗脱,采用正离子模式,多反应监测方式进行定性和定量分析。该方法能进行快速分析,检测范围更广,较之前灵敏、准确。运用上述实验方法分别对杭州市两处水库水样中的MC进行检测,结果显示分别有3种和8种MC被检测出来。
液相色谱质谱联用技术较好地解决了单纯使用高效液相色谱仪检测时的部分问题,其缺点是缺少各种MC的标准物质,且实验成本较高。
1.3 毛细管电泳法
毛细管区带电泳、胶束电动毛细管与紫外、荧光、质谱检测器串接也被用于微囊藻毒素的检测,该方法具有自动化、检测样品量多、用量少等优点,且此方法具有柱上富集的功能。同生物法相比,该方法自动化程度更高,且在实验中不使用放射性物质。但是其灵敏度没有HPLC高,检测下限仅为l mg/L[30]。应用激光诱导荧光检测器可提高检测灵敏度[31],但是还需进行进一步的研究和探索。Siren等[32]在对蓝藻肝毒素进行分离、纯化和鉴别时使用了毛细管电泳和电淋洗质谱技术。首先利用HPLC方法把受试化合物进行预分离,然后在胶束电动色谱(MECC)方法下将这些受试物逐一分离,最后用离线电质谱对分离后的微囊藻毒素进行鉴别和高灵敏度纯化验证实验,得出MC的检出限低于1 μg/L。此方法的特点是样品用量小、分离效率高、分析时间短且柱上富集功能强。但由于重现性差,且缺少MC标准物质,所以目前毛细管电泳法还暂时不能作为水体检测藻类毒素的常规手段。
1.4 气相色谱法
GC–MS联用技术主要用于MC总量测定,其检出限可达到ng/L级。Tsuji[33]等发明了2-甲基-3-甲氨基-4-氨基铬酸(MMPB)检测方法,利用碘酸钠和高锰酸钾对提取后样品进行氧化的原理。MMPB是与MC共同氧化的中间产物,利用GC测定MMPB的含量即可间接获得MC的总含量,该方法的缺点是只能测MC的总量,无法定量不同的MC异构体。Harada等[34]实现了利用臭氧分解产生MMPB的方法,将存在于甲醇溶液中的藻细胞在–78℃的低温条件下直接进行臭氧分解,然后用气相色谱–质谱(GC–MS)进行检测,30 min内完成整个检测过程,其检出限能达到ng/L级。GC–MS的优点是能够灵敏、快速、准确地对痕量MC进行定量,其缺点是仪器设备价格昂贵、不易普及,技术含量高,操作复杂,在缺少标准品的情况下不能对微囊藻毒素进行定性定量,前处理过程也较为复杂,各实验室的条件和检测程序千差万别,对数据的分析和判断都有影响。现阶段的研究显示,在高灵敏度下进行检测同样也会对实验的准确性产生影响,样品很快会被实验用品、试剂甚至外界的空气等污染,而且由于MC本身具有毒性,其气化进入空气中也会对人类造成危害。
1.5 薄层色谱法
薄层色谱法(TLC)是将固定相涂布在平板(玻璃板、铝箔等)上,点样后在合适的流动相条件下进行洗脱,平板上不同组分的毒素就会逐渐分离。此方法是利用MC及其衍生物与有色或荧光物质发生反应的一种检测技术,目前,它正向联用的方向发展,如薄层色谱–气相色谱–质谱联用(TLC–GC–MS),或薄层色谱–红外光谱(TLC–IR)等的联用。Pelander等[35]使用N,N-二甲基-1,4-苯二胺二氯化物(N,N-DPDD)可以优化TLC法检测MCs,能够达到WHO规定的1 μg/L的检测极限。薄层色谱的优点是具有较高的灵敏度及分辨率,可进行快速分离,易于操作,多个样品的分离可同时进行,样品预处理简单。但由于薄层色谱法在检测水体中藻毒素时无法达到高检出限、准确定量等实验的需求,至今未被广泛应用于实际生活中,只能用于小规模的实验室科研工作。
2 生物法
生物法是检测MC实施最早,最普遍的方法。它通过急性毒素实验对MC进行定性和半定量检测。它可快速直观检测到新的不同藻毒素,但消耗的毒素量较多,且灵敏度和专一性都不高,不能运用该方法对藻毒素进行准确地定量测定,更不能对不同的同分异构体进行分离和辨认。且当样品中神经毒素和MC同时存在时,肝毒素的毒性就不能显示。用口腔灌喂法或鼠腹腔注射法是判断MC的毒性的一种传统方法。目前,已建立许多种生物系统来监测MC,有利用无脊椎动物对毒性评价进行研究,也有利用细菌进行毒性试验研究[36]。由于MCs能阻碍费舍尔弧菌或明亮发光杆菌等一些发光菌发光,Lawton[37]等利用这一特性检测了MC–LR和含有5种MCs的蓝藻样品,其LD50的检测范围为0.02~0.46 μg/mL。Gehringer等[38]运用植物试验来检测水体中的MC,实验中把家独行菜作为检测的物质,将秧苗持续放置在在1 μg/L MC–LR中2~6 d时,根和叶部都出现了不同的变化。
3 生物化学法
在MC的检测方法中,生物化学法也是开展比较早的方法。它在检测MC对人或其它生物的生理活性方面有独特的优势。生物化学的方法主要分为蛋白磷酸酶抑制检测法(PPIA)和酶联免疫吸附检测法(ELISA)。
MC对蛋白磷酸酶活性具有专一性抑制的特性,PPIA就是依据酶活性抑制和MC含量之间的相关关系来测定毒素含量的一种方法[39]。常用的方法包括微量比色法和同位素标记法。该法具有较高的灵敏度,检测下限可以达到1 ng/mL~1 pg/mL。PPIA方法的缺陷是待测样品处理需要较高技术,而且常出现MC含量高估计误差。目前,PPIA常与色谱(如HPLC)结合使用,主要用于水样和动物组织样品中痕量MC的检测。
酶联免疫法的原理是用制备的MC单克隆或多克隆抗体,通过免疫显色反应来测定MC含量。该方法多用于对MC的总量进行测定,最近ELISA方法得到了改进和发展,现已广泛用于各种动植物组织细胞样品、实验室蓝藻培养物和蓝藻水华水样中MC含量的测定。ELISA方法具有高灵敏度,通常能检测到1 ng/mL,有些能达到1 pg/mL,但由于其较低的选择性,对MC异构体尚不能进行区分,只能测定总MC含量。因此,目前ELISA一般在水样和动物组织样品中微量MC的检测及MC快速检测时使用。
表1中列出了8种常用的MC检测方法比较结果。
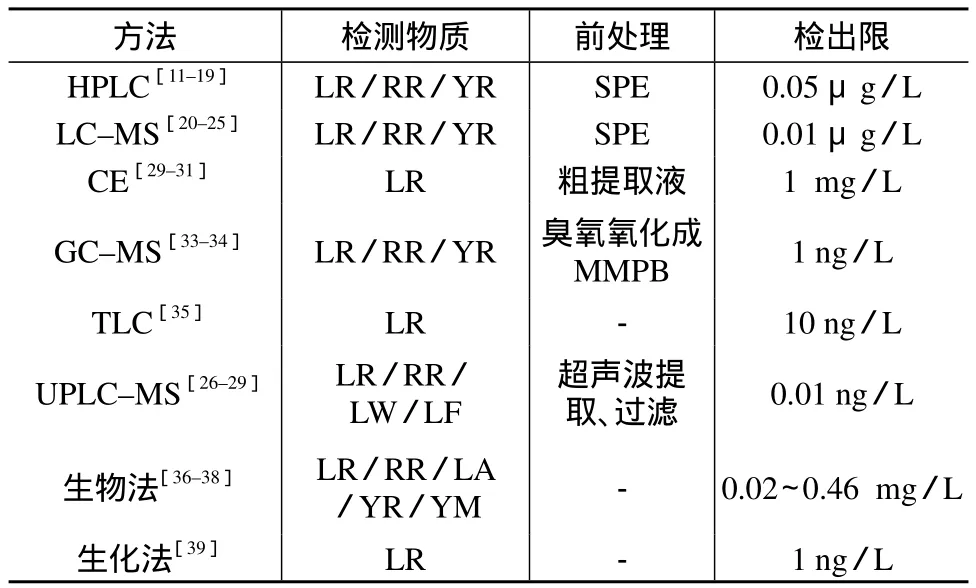
表1 8种常用分析检测方法比较结果
4 展望
目前,MC检测技术种类繁多,不同的分析检测方法利用自身的特点都在追求着多方向的发展。但是因为各种分析技术及各种条件都存在差异,对MC的测定结果没有达成统一。现在,许多研究机构致力于对藻毒素的检测和分离方法的研究,他们提出了各种不同的思路和方法,但均有各自的优势和劣势。生物法虽然简单快速,但灵敏度低,没有特异性;色谱–质谱技术虽然能够精确定量,但仪器昂贵,技术要求高;生化检测虽然灵敏度较高,且经济实惠,检测比较迅速,但目前仍无法解决的是商用试剂盒广泛供应问题。在定量检测和分析MC时,高效液相色谱法作为最经典可靠的技术,其检出限可达到ng及以下水平,同时具有选择性好,结果可靠等优点。所以开展有关HPLC检测MC的方法学研究,用以对各种MC的同分异构体进行分离,尽快研制MC标准物质,从而为进一步发现并鉴定新毒素,分析其结构特性,为对水质和生物体中MC的研究和控制消除提供更强有力的依据。
[1] Rositano J J,et al.Water Res,200l,35(1): 23–32.
[2] 闫海,等.环境科学学报,2004,24(2): 355–359.
[3] Carmicbael WW,et al.Toxicon,1988,26: 971–973
[4] Mekebri A,et al.Journal of Chromatography A,2009,1 216: 3 147–3 155.
[5] Schripsema J, et al. Magn Reson Chem, 2002,40:614–617.
[6] Goldberg J,et al. Nature,1995,376: 745–753
[7] 聂晶晶,等.中国环境检测.2007,23(2): 44–48.
[8] Pelander A,et a1. Water Res,2000,34 (10): 2 643–2 652.
[9] Tsuji K,et al.Toxicon,200l,39 (5): 687–692.
[10] Vasas G,et al.Biochem Biophys Methods,2006,66: 87–97.
[11] 韩志国,等.中国环境监测,2001,17(6): 35–39.
[12] Harada K I,et a1. Phycologia,1996,35(Supple): 36–41.
[13] 周志刚.铁氧体磁性材料[M].北京:科学出版社,1 981: 6–73.
[14] Esme L Purdie,et al. Toxicon,2009,54: 887–890.
[15] 房祥军,等.中国食品学报,2011,11(2): 209–215
[16] Christine Edwards,et al. Journal of Chromatography A,2010,1 217: 5 233–5 238.
[17] Moreno I M,et al. Journal of Chromatography A,2005,1 080(2): 199–203
[18] 闫海,等.生态学报,2002,22(11): 1 968–1 975.
[19] H S Lee,et a1.Journal of Chromatography A,1999,848: 179–184.
[20] Carmela Dell Aversano,et al. Journal of Chromatography A,2004,1 028(1): l55–164.
[21] WHO.Cyanobacterial toxins: microcystin—LR in drinking water.background document for the development of WHO guidelines for drinking water quality.Geneva: WHO,2003: 4–5.
[22] 虞锐鹏,等.分析化学简报,2003,32(12): 1 462–1 464.
[23] Werawan Ruangyuttikarn,et al. Journal of Chromatography B,2004,800: 315–319.
[24] 张昱,等.中国给水排水,2005,21(4): 94–96.
[25] 王超,等.色谱,2011,29(3): 212–216.
[26] 王静,等.色谱,2006,24(4): 335–338.
[27] Ortelli D,et al. Anal chimica acta,2008,617: 230–237.
[28] 茅海琼,等.中国环境监测,2009,25(6): 19–22.
[29] 张明,等.色谱,2012,30(1): 51–55.
[30] Vasas G,et al. Electrp Horesis,2004,25(1): l08–115.
[31] Li P C H,et al. Mar Pollut Bull,l999,39: 250–254.
[32] Siren H,et al. J Chromatogra A,1999,839: 203–215.
[33] Tsuji K,et al. Environ Sci Technol,1994,28: 173–177.
[34] Harada K I,et al. Toxicon,1996,34: 701–710.
[35] Pelander A,et al. Water Research,2000,34(10): 2 643–2 652.
[36] Sturgeon S A,et al. Biochimica & Biophysica Acta,1999,1 454(3): 227–235.
[37] Lawton L A,et a1. Lett Appl Microbiol,1990,11: 205–207.
[38] Gehringer Michelle M,et a1. Toxicon,2003,41(7): 871–876.
[39] Bouacha N,et al. Food Chem Toxicol,2002,40 (11): 1 677–1 683.
猜你喜欢
化工设计通讯(2022年10期)2022-12-31
波谱学杂志(2022年2期)2022-06-14
考试与评价·高二版(2021年3期)2021-09-10
数学物理学报(2020年5期)2020-11-26
天然产物研究与开发(2018年8期)2018-09-10
中国交通信息化(2017年9期)2017-06-06
公民与法治(2016年14期)2016-05-17
工业设计(2016年11期)2016-04-16
现代检验医学杂志(2015年1期)2015-02-06
河南科技(2014年22期)2014-02-27

- 化学分析计量的其它文章
- X–射线荧光光谱法测定白云石中主次量组分