
Mpro-C蛋白三维结构域交换的机理:来自分子模拟的线索
2012-12-11 09:12黄永棋刘志荣
物理化学学报 2012年10期
黄永棋 康 雪 夏 斌,5 刘志荣,*
(1北京大学化学与分子工程学院,北京100871;2北京大学分子动态与稳态结构国家重点实验室,北京分子科学国家实验室,北京100871;3北京大学定量生物学中心,北京100871;4北京核磁共振中心,北京100871; 5北京大学生命科学学院,北京100871)
Mpro-C蛋白三维结构域交换的机理:来自分子模拟的线索
黄永棋1,2,3康 雪1,4夏 斌1,4,5刘志荣1,2,3,*
(1北京大学化学与分子工程学院,北京100871;2北京大学分子动态与稳态结构国家重点实验室,北京分子科学国家实验室,北京100871;3北京大学定量生物学中心,北京100871;4北京核磁共振中心,北京100871;5北京大学生命科学学院,北京100871)
SARS冠状病毒主蛋白酶(Mpro)在病毒的蛋白酶切过程中发挥着重要作用.Mpro的晶体结构显示它存在两种形式的二聚体:一种是发生三维结构域交换的形式,另一种是非交换的形式.Mpro的C端结构域(Mpro-C)单独表达时也能形成与全长Mpro类似的三维结构域交换二聚体.三维结构域交换通常发生在蛋白质的表面,但Mpro-C的结构域交换却发生在疏水核心.在本文中,我们利用分子动力学模拟及三维结构域交换预测算法研究了Mpro-C中被高度埋藏的核心螺旋片段发生交换的机理.我们发现基于结构与基于序列的已有算法都不能正确预言出Mpro-C和Mpro中发生结构域交换的铰链区位置.分子模拟结果表明Mpro-C中的交换片段在天然态下埋藏得很好,但在变性单体中则会被释放并暴露在外面.因此,在完全或部分解折叠状态下交换片段的打开有助于促进单体间的相互作用及结构域交换二聚体的形成.
SARS冠状病毒;主蛋白酶;分子模拟;结构域交换;蛋白质-蛋白质相互作用;蛋白质解折叠
1 Introduction
Three-dimensional(3D)domain swapping is one of the mechanisms by which protein complexes are formed.In a 3D domain swapping process,protein monomers exchange identical domains or structural elements during oligomerization.1Great efforts have been devoted to understanding the structural features and biological functions of 3D domain swapping. There is evidence that 3D domain swapping mediates amyloid fibril formation.2-5Furthermore,3D domain swapping is a mechanism underlying the evolution of protein complexes6-8and promotes signal transduction within large protein complexes.9,10
Analyses of the structures of domain-swapped complexes show that there is no general features(including size,sequence,and secondary structure)of the exchanged regions.11However,in most experimental observations of domainswapped oligomers,the swapped regions locate to the surface of protein 3D structures,i.e.,the swapped regions are not wrapped by other structural elements,11-13suggesting that native topology is a general determinant of 3D domain swapping.14Topology-based methods have achieved some success in reproducing the 3D domain-swapped structures or locating the hinge loop regions based on monomer structures,14-18further supporting the role of structural topology in 3D domain swapping.The swapping of surface regions may be promoted by the local unfolding of proteins,whereby the swapped regions are released and are able to form extensive intermolecular interactions between monomers.12,19
SARS coronavirus main protease(Mpro)is a key enzyme involved in the extensive proteolytic processing of the virusʹ polyproteins and is an attractive target for anti-SARS drug design.Crystal structures of Mproreveal that the enzyme contains two domains(an N-terminal domain and a C-terminal domain) and exists as a homodimer in which a domain-swapped form20and a non-swapped form21have been observed.The isolated C-terminal domain(Mpro-C)alone also forms a domainswapped structure(Fig.1A)identical to that formed by the full-length protein.22Unlike previously observed 3D domainswapped structures,Mprois unique in that the swapped region (T201−N214,α1-helix)locates to the core rather than the surface of a folded domain.To date,the swapping mechanism of Mprohas not been solved.
In this work,we used molecular dynamics(MD)simulations and 3D domain-swapping predictions to investigate how a highly buried core helix in a helix bundle structure of Mpro-C is swapped.
2 Materials and methods
A series of MD simulations were performed on the Mpro-C monomer and the 3D domain-swapped dimer.Initial conformations for the simulations were taken from the solution structure of Mpro-C monomer(PDB 2K7X)22and the crystal structure of Mpro-C dimer(PDB 3EBN).22The duration of the simulations was 50 ns at 298 K and 100 ns at 498 K for both monomer and dimer.
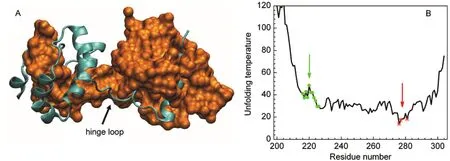
Fig.1 (A)Structure of domain-swapped Mpro-C dimer(PDB 3EBN),to clearly show the swapped core helix,one chain is shown by a cyan ribbon and the other by an orange surface.(B)H-Predictor prediction for Mpro-C,red stars indicate the predicted hinge loop regions; green dots indicate the experimentally observed hinge loop regions.
Simulations were performed using the GROMACS 4.5.1 program23,24and the OPLS-AA/L force field.25Water molecules were modeled by the SPC/E representation.26Each of the starting conformations was placed in the center of a cubic water box with a distance of at least 1.0 nm from the box edge.Peri-odic boundary conditions were used and Na+ions were added to neutralize the net charges.The long-range electrostatic interactions were treated with the particle mesh Ewald method.27The bond lengths were fixed by using the LINCS algorism,28and a time step of 2 fs was used.After relaxation by 1000 steps of the steepest-descent energy minimization,each system was equilibrated at 298 K by 100 ps under an NVT ensemble and further equilibrated for 200 ps at constant pressure(1×105Pa). Production simulations were performed at constant temperature(298 or 498 K)and pressure(1×105Pa).Coordinates were saved every 5 ps.V-rescale29and Parrinello-Rahman30were used to couple the system to the simulation temperature and pressure with coupling constants of 0.1 and 2.0 ps,respectively.
We monitored the contacts between the two chains in the domain-swapped dimer to investigate the intermolecular interactions during simulations.An intermolecular contact was considered to be formed between two residues(one contributed by each chain)when the distance between any two non-H atom pairs was≤0.45 nm.Secondary structure analysis was assigned using the DSSP program.31Molecular graphics images were created using the VMD program.32
3 Results
3.1 Hinge loop prediction
The hinge loop is the region linking the swapped domain and the main domain.Because of the special role of the hinge loop playing in a swapping process,much effort has been devoted to the development of methods for locating the hinge loop regions based on monomeric structures and/or sequences. Two different methods were used here to predict the location of the hinge loop or the probability of domain swapping in Mproand Mpro-C.The first method was H-Predictor,14which is based on the 3D structure of a protein and calculates the effective unfolding temperature for each residue using a simple contactbased potential for enthalpy and a graph theory-based estimation for entropy.Residues with the lowest effective unfolding temperature will have the highest probability of being in the hinge region.Fig.1B shows the results for Mpro-C predicted by H-Predictor.The predicted hinge loop regions are the C-terminal loop(ca 276-281)for Mpro-C(Fig.1B)or the loop linking the C-terminal and N-terminal domains(ca 186-193)for Mpro(data not shown).Neither of the predicted hinge loops coincides with the hinge loop determined experimentally(ca 214-226).The second method is 3dswap-pred.33Unlike H-Predictor,3dswap-pred predicts the probability of domain swapping based on protein sequences using the random forest approach.3dswap-pred succeeded in predicting Mproto be a domain-swapped protein but incorrectly predicted Mpro-C to be a non-domain-swapped protein.3dswap-pred does not provide the location of the possible hinge loop.The discrepancy between predictions and the experimental results suggests that the domain swapping in Mpro-C is not conventional.
3.2 Flexibility of the domain-swapped dimer
The dynamic nature of domain-swapped complexes has been observed in previous simulations.34-37For Mpro-C,the simulated backbone root mean square fluctuation(RMSF)at 298 K shows that its structural flexibility is increased in the transition from monomer to domain-swapped dimer(Fig.2A).The increased flexibility of the domain-swapped dimer is consistent with experimental B-factors(Fig.2B).It is noted that the hinge loop does not locate to the region with the highest flexibility. This may be the reason why the structure-based method H-Predictor failed to correctly locate the hinge loop in Mpro-C.The flexibility of the domain-swapped dimer can also be revealed by investigating intermolecular contacts in the open interface. The open interface in the Mpro-C dimer is mainly formed between the two hinge loops and between the two main domains. During simulations,contacts between the two hinge loops (Fig.2C)and between the two main domains(Fig.2D)fluctuated to a certain extent,indicating the dynamic feature of the open interfaces.
3.3 Compacting of the unfolded monomer and dimer
The unfolding of a protein(fully or partially)and opening of the swapped region has been proposed as a mechanism for domain swapping in several proteins.12,19Unlike previously studied proteins,the swapped region of Mpro-C locates to the core of a folded structure and is wrapped by four other helices (Fig.1A).It is currently unclear whether the swapped region of Mpro-C is released and exposed in the unfolded state.To address the structural properties of unfolded Mpro-C,we performed simulations at 498 K.Simulation trajectories indicate that the unfolding process initiates from the C-terminal helix, which is consistent with the experimental observation that the C-terminal helix exhibits the lowest stability and unfolds in the presence of 2.5 mol·L-1urea.38Figs.3A and 3B indicate that the Mpro-C monomer unfolds within about 40 ns.The unfolded monomer retains a compact structure and the average radius of gyration of the unfolded conformations(40-100 ns)is only (0.13±0.06)nm greater than the native folded state(Fig.3C).
Unfolding of the Mpro-C dimer takes longer than that of the monomer and the CαRMSD does not reach a plateau within 100 ns of simulation(Fig.3D).The partially unfolded dimer is also compact as revealed by intermolecular contacts between the two monomers(Fig.3E)and the radius of gyration (Fig.3F).Extensive intermolecular contacts and a decrease in the radius of gyration indicate that the partially unfolded dimer forms a united molten structure rather than being two individual components.
3.4 Opening of the swapped helix in the unfolded monomer
Although the unfolded Mpro-C monomer is compact,solvent accessible surface area(SASA)indicates that the swapped region is released and exposed in the unfolded state.In the native state,SASA of the swapped region is 4.75 nm2,while in the unfolded state(40-100 ns),SASA increases to(10.57±2.04)nm2. Dividing SASA into hydrophobic and hydrophilic,we find that a great part of the increased SASA is hydrophobic,from 2.11 nm2in the native state to(7.00±1.48)nm2in the unfolded state (Fig.4A).Hydrophilic SASA only increases moderately (Fig.4B).Snapshots of simulations clearly show that the swapped region is exposed in the unfolded state(Fig.4C).Our results indicate that regions with the lowest stability may not possess the highest propensity of being swapped and regions that are highly buried but exposed in the unfolded state may also be swapped.Therefore,more factors are needed to be considered to further improve the hinge loop prediction methods.
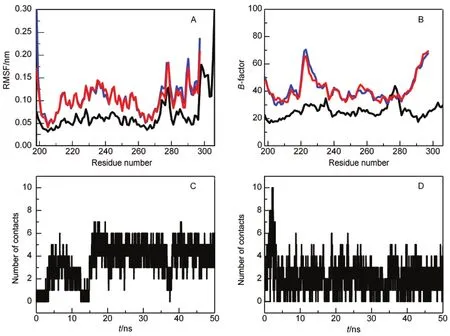
Fig.2 Comparison of the flexibility between the monomer and the domain-swapped dimer of Mpro-C(A)simulated backbone RMSF and(B)experimental B-factor for the monomer and the dimer,the monomer is indicated by a black line and the two chains in the dimer are indicated by red and blue lines.(C)Number of contacts between the hinge loops and(D)number of contacts between the main domains in the dimer during simulations
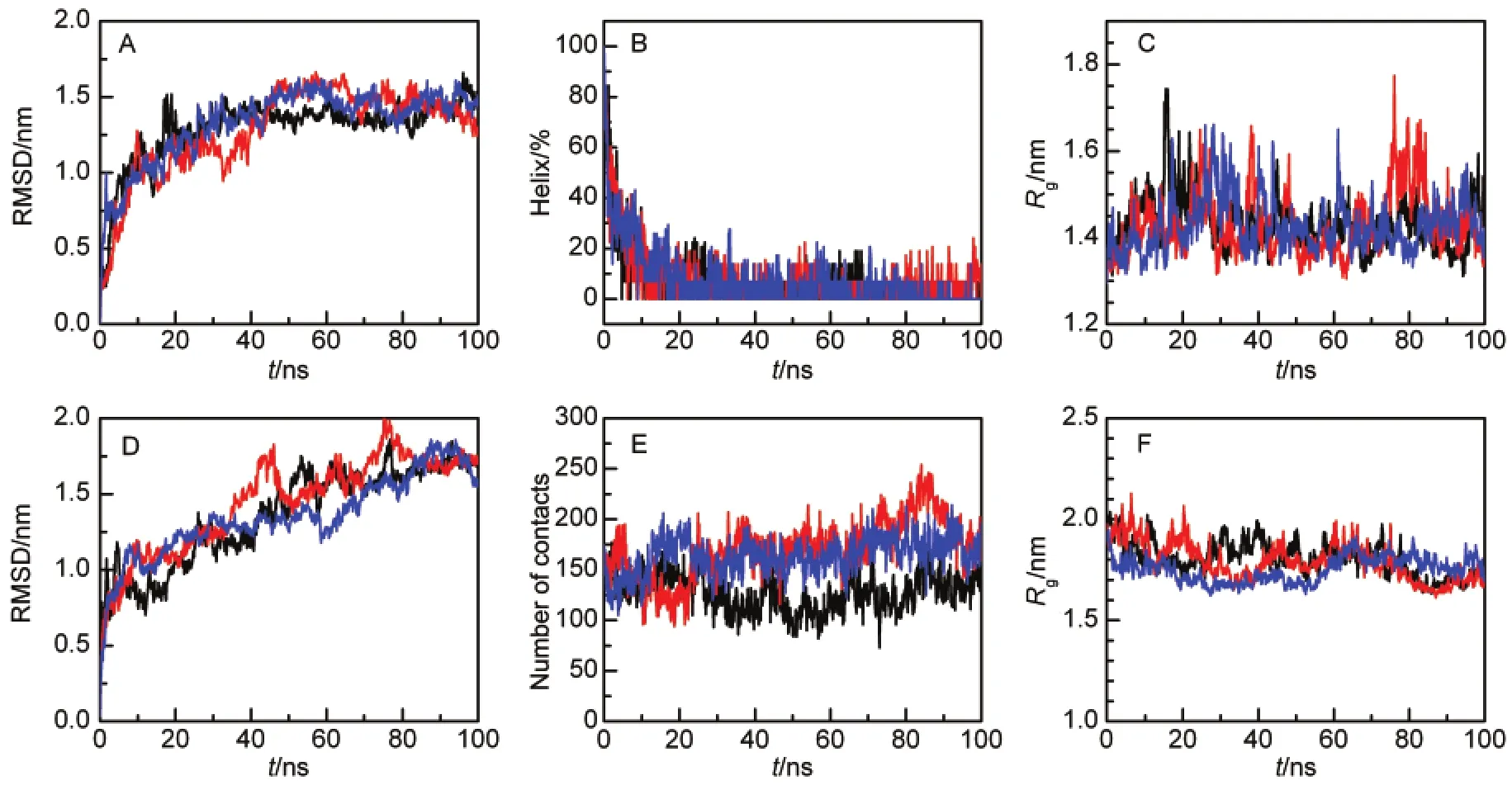
Fig.3 Unfolding simulations of Mpro-C(A-C)monomer and(D-F)domain-swapped dimer(A)CαRMSD relative to the native monomer structure;(B)percentage of residues in the α-helical structure;(C)radius of gyration of all protein atoms;(D)CαRMSD relative to native dimer structure;(E)number of contacts between the two monomers;(F)radius of gyration of all protein atoms.Three simulation trajectories are shown.
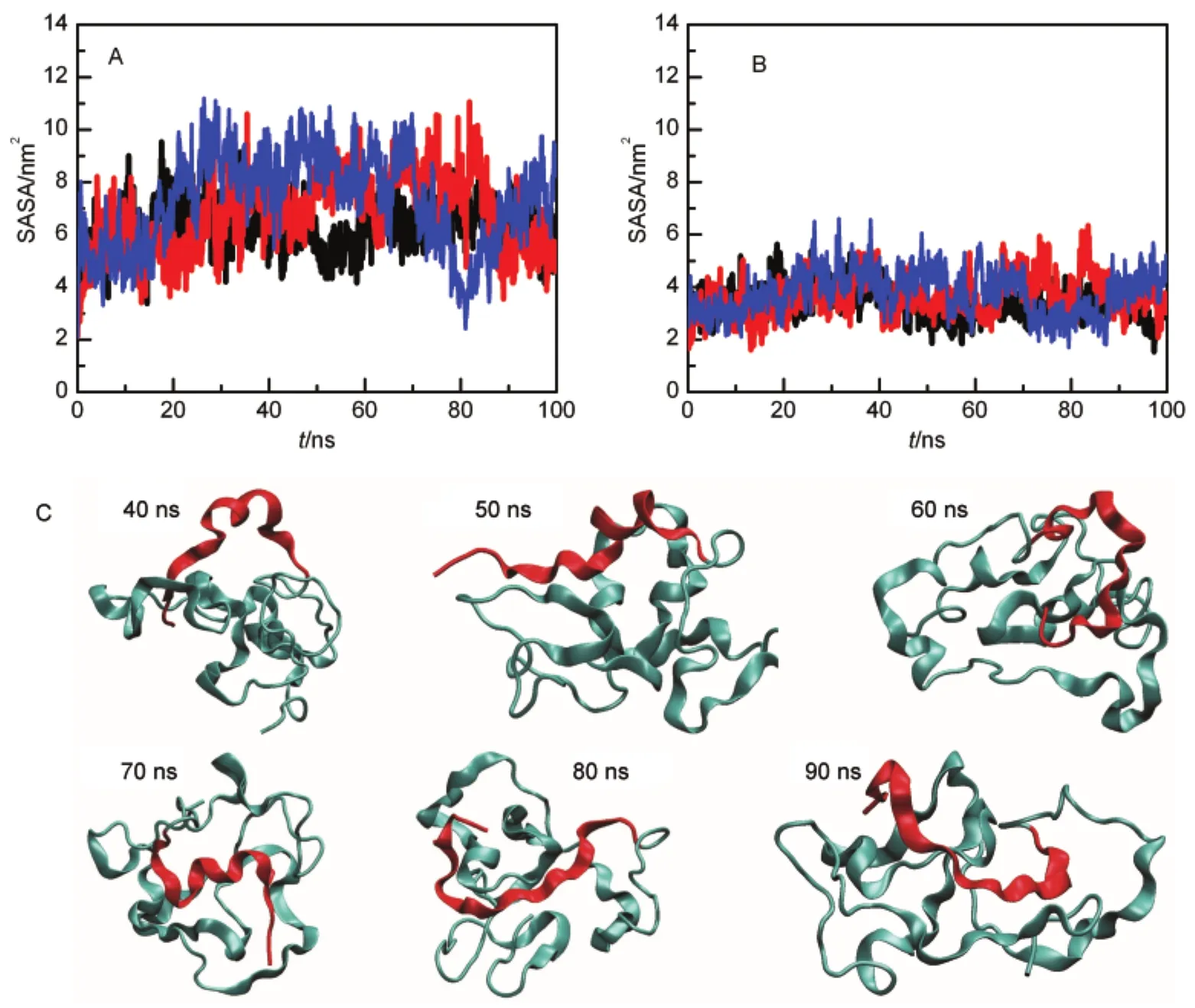
Fig.4 SASAof the swapped helix during unfolding simulations of the monomer(A)hydrophobic SASA,(B)hydrophilic SASA,three simulation trajectories are shown;(C)representative unfolded conformations of the Mpro-C monomer in one trajectory.The swapped region is shown in red.
4 Discussion
As a mechanism for forming protein complexes,3D domain swapping has been observed in about 10%of structural classification of protein(SCOP)fold types and 5%of SCOP families.13Extensive distribution of 3D domain swapping in the protein structure space indicates that 3D domain swapping plays important biological functions.To clarify how 3D domainswapped structures are formed,many studies have been conducted to investigate the mechanism of 3D domain swapping. A general picture has been obtained for some proteins,whereby the swapping process starts from the fully or partially unfolded state.12,19,39The unfolding of a protein and the opening of the swapped region promote interactions between the swapped region of one monomer and the main domain of another monomer,and thus increase the probability of forming 3D domain-swapping complexes.It is expected that mutations which destabilize a monomer will increase the population of the domain-swapped form,e.g.,cyanovirin-N.40
The opening of swapped regions in the unfolded state has been observed in several proteins,e.g.,p13suc1,41ribonuclease A,42GB1,43FIS,44and SH3 domain.15However,simulations with GB1 showed that unfolding may be not necessary in the swapping process,and that the conformational changes of monomers are tightly coupled to the swapping process.45NMR measurements of the isolated EC1 domain of type II cadherin-8 identified monomers with exposed swapped regions, and it was proposed that swapping of the EC1 domain of cadherin-8 proceeds via a selected-fit mechanism.46However,later studies by Sivasankar et al.,47suggested that cadherin dimerization proceeds via an induced fit mechanism.We noted that the GB1 domain and the EC1 domain of cadherin-8 share one common structural feature that the swapped region is exposed at the surface even in the folded monomer.This enables monomers to form intermolecular interactions between the swapped region of one monomer and the main domain of the other monomer,and then to undergo subsequent conformational changes to form the final domain-swapped dimer.
In contrast to the GB1 and EC1 domains of cadherin-8,the swapped regions of Mpro-C and Mprolocate to the core of a folded structure.Weak interactions that form during encounters between two folded monomers may not enable the folded structures to deform and release the swapped regions.Recently, Kang et al.38studied the swapping kinetics and thermodynamics of Mpro-C and found that unfolding of the C-terminal α5-helix is required for Mpro-C to form domain-swapped dimer.It is proposed that unfolding of the C-terminal helix converts Mpro-C into an active state,which promotes Mpro-C to form an intermediate dimer.The N-terminal α1-helices are exchanged in the intermediate dimer.Exposure of the α1-helix in the unfolded monomer may facilitate α1-helix from one Mpro-C monomer to form extensive hydrophobic interactions with the other Mpro-C monomer and so promote the formation of domainswapped dimer.
5 Conclusions
By using 3D domain-swapping predictions and molecular dynamics simulations,we studied the swapping mechanism of Mpro-C and Mpro.We found that both structure-based and sequence-based methods failed to predict the hinge loop location in Mpro-C and Mpro.We then performed extensive molecular dynamics simulations to investigate the structural properties of the unfolded monomer and the domain-swapped dimer.We found that although the swapped region was buried in the native state,it was exposed in the unfolded monomer.Our results suggest that exposure of the swapped region in the unfolded state may promote interactions between monomers and the formation of domain-swapped structures in Mpro-C and Mpro.
(1) Bennett,M.J.;Schlunegger,M.P.;Eisenberg,D.Protein Sci. 1995,4,2455.doi:10.1002/pro.v4:12
(2) Nelson,R.;Eisenberg,D.Curr.Opin.Struct.Biol.2006,16, 260.doi:10.1016/j.sbi.2006.03.007
(3) Liu,C.;Sawaya,M.R.;Eisenberg,D.Nat.Struct.Mol.Biol. 2011,18,49.doi:10.1038/nsmb.1948
(4) Žerovnik,E.;Stoka,V.;Mirtič,A.;Gunčar,G.;Grdadolnik,J.; Staniforth,R.A.;Turk,D.;Turk,V.FEBS J.2011,278,2263. doi:10.1111/j.1742-4658.2011.08149.x
(5) Bennett,M.J.;Sawaya,M.R.;Eisenberg,D.Structure 2006, 14,811.doi:10.1016/j.str.2006.03.011
(6) Bergdoll,M.;Eltis,L.D.;Cameron,A.D.;Dumas,P.;Bolin,J. T.Protein Sci.1998,7,1661.doi:10.1002/pro.v7:8
(7) DʹAlessio,G.Prog.Biophys.Mol.Biol.1999,72,271.doi: 10.1016/S0079-6107(99)00009-7
(8) Hadjithomas,M.;Moudrianakis,E.N.Proc.Natl.Acad.Sci.U. S.A.2011,108,13462.doi:10.1073/pnas.1108649108 (9)Schymkowitz,J.W.H.;Rousseau,F.;Wilkinson,H.R.; Friedler,A.;Itzhaki,L.S.Nat.Struct.Biol.2001,8,888.doi: 10.1038/nsb1001-888
(10) Shi,Q.;Maruthamuthu,V.;Li,F.;Leckband,D.Biophys.J. 2010,99,95.doi:10.1016/j.bpj.2010.03.062
(11) Liu,Y.S.;Eisenberg,D.Protein Sci.2002,11,1285.doi: 10.1110/ps.0201402
(12) Gronenborn,A.M.Curr.Opin.Struct.Biol.2009,19,39.doi: 10.1016/j.sbi.2008.12.002
(13) Huang,Y.;Cao,H.;Liu,Z.Proteins 2012,doi:10.1002/ prot.24055.
(14) Ding,F.;Prutzman,K.C.;Campbell,S.L.;Dokholyan,N.V. Structure 2006,14,5.doi:10.1016/j.str.2005.09.008
(15) Yang,S.C.;Cho,S.S.;Levy,Y.;Cheung,M.S.;Levine,H.; Wolynes,P.G.;Onuchic,J.N.Proc.Natl.Acad.Sci.U.S.A. 2004,101,13786.doi:10.1073/pnas.0403724101
(16) Chen,Y.W.;Dokholyan,N.V.J.Mol.Biol.2005,354,473.doi: 10.1016/j.jmb.2005.09.075
(17) Chahine,J.;Cheung,M.S.Biophys.J.2005,89,2693.doi: 10.1529/biophysj.105.062679
(18) Song,G.;Jernigan,R.L.Proteins 2006,63,197.doi:10.1002/ prot.20836
(19) Rousseau,F.;Schymkowitz,J.W.H.;Itzhaki,L.S.Structure 2003,11,243.doi:10.1016/S0969-2126(03)00029-7
(20) Zhang,S.;Zhong,N.;Xue,F.;Kang,X.;Ren,X.;Chen,J.;Jin, C.;Lou,Z.;Xia,B.Protein Cell 2010,1,371.doi:10.1007/ s13238-010-0044-8
(21)Yang,H.;Yang,M.;Ding,Y.;Liu,Y.;Lou,Z.;Zhou,Z.;Sun, L.;Mo,L.;Ye,S.;Pang,H.;Gao,G.F.;Anand,K.;Bartlam, M.;Hilgenfeld,R.;Rao,Z.Proc.Natl.Acad.Sci.U.S.A.2003, 100,13190.doi:10.1073/pnas.1835675100
(22) Zhong,N.;Zhang,S.N.;Xue,F.;Kang,X.;Zou,P.;Chen,J.X.; Liang,C.;Rao,Z.H.;Jin,C.W.;Lou,Z.Y.;Xia,B.Protein Sci. 2009,18,839.
(23) Berendsen,H.J.C.;van der Spoel,D.;van Drunen,R.Comp. Phys.Commun.1995,91,43.doi:10.1016/0010-4655(95) 00042-E
(24) Hess,B.;Kutzner,C.;Spoel,D.v.d.;Lindahl,E.J.Chem. Theory Comput.2008,4,435.doi:10.1021/ct700301q
(25) Kaminski,G.A.;Friesner,R.A.;Tirado-Rives,J.;Jorgensen,W. L.J.Phys.Chem.B 2001,105,6474.doi:10.1021/jp003919d
(26) Berendsen,H.J.C.;Grigera,J.R.;Straatsma,T.P.J.Phys. Chem.1987,91,6269.doi:10.1021/j100308a038
(27) Darden,T.;York,D.;Pedersen,L.J.Chem.Phys.1993,98, 10089.doi:10.1063/1.464397
(28) Hess,B.;Bekker,H.;Berendsen,H.J.C.;Fraaije,J.G.E.M. J.Comput.Chem.1997,18,1463.doi:10.1002/(ISSN) 1096-987X
(29) Bussi,G.;Donadio,D.;Parrinello,M.J.Chem.Phys.2007, 126,014101.doi:10.1063/1.2408420
(30) Parrinello,M.;Rahman,A.J.Appl.Phys.1981,52,7182.doi: 10.1063/1.328693
(31) Kabsch,W.;Sander,C.Biopolymers 1983,22,2577.doi: 10.1002/(ISSN)1097-0282
(32) Humphrey,W.;Dalke,A.;Schulten,K.J.Mol.Graph.1996,14, 33.doi:10.1016/0263-7855(96)00018-5
(33)Shameer,K.;Pugalenthi,G.;Kandaswamy,K.K.;Sowdhamini, R.Protein Pept.Lett.2011,18,1010.doi:10.2174/ 092986611796378729
(34) Merlino,A.;Vitagliano,L.;Ceruso,M.A.;Mazzarella,L. Biophys.J.2004,86,2383.doi:10.1016/S0006-3495(04) 74295-2
(35) Kundu,S.;Jernigan,R.L.Biophys.J.2004,86,3846.doi: 10.1529/biophysj.103.034736
(36) Merlino,A.;Ceruso,M.A.;Vitagliano,L.;Mazzarella,L. Biophys.J.2005,88,2003.doi:10.1529/biophysj.104.048611
(37) Cailliez,F.;Lavery,R.Biophys.J.2006,91,3964.doi:10.1529/ biophysj.106.087213
(38) Kang,X.;Zhong,N.;Zou,P.;Zhang,S.;Jin,C.;Xia,B.Proc. Natl.Acad.Sci.U.S.A.2012,109,14900.doi:10.1073/ pnas.1205241109.
(39) Liu,L.;Byeon,I.J.;Bahar,I.;Gronenborn,A.M.J.Am.Chem. Soc.2012,134,4229.doi:10.1021/ja210118w
(40) Barrientos,L.G.;Louis,J.M.;Botos,I.;Mori,T.;Han,Z.; OʹKeefe,B.R.;Boyd,M.R.;Wlodawer,A.;Gronenborn,A.M. Structure 2002,10,673.doi:10.1016/S0969-2126(02)00758-X
(41) Rousseau,F.;Schymkowitz,J.W.;Wilkinson,H.R.;Itzhaki,L. S.J.Biol.Chem.2004,279,8368.
(42) Esposito,L.;Daggett,V.Biochemistry 2005,44,3358.doi: 10.1021/bi0488350
(43) Byeon,I.J.L.;Louis,J.M.;Gronenborn,A.M.J.Mol.Biol. 2004,340,615.doi:10.1016/j.jmb.2004.04.069
(44)Topping,T.B.;Hoch,D.A.;Gloss,L.M.J.Mol.Biol.2004, 335,1065.doi:10.1016/j.jmb.2003.11.013
(45) Malevanets,A.;Sirota,F.L.;Wodak,S.J.J.Mol.Biol.2008, 382,223.doi:10.1016/j.jmb.2008.06.025
(46) Miloushev,V.Z.;Bahna,F.;Ciatto,C.;Ahlsen,G.;Honig,B.; Shapiro,L.;Palmer,A.G.Structure 2008,16,1195.doi: 10.1016/j.str.2008.05.009
(47) Sivasankar,S.;Zhang,Y.;Nelson,W.J.;Chu,S.Structure 2009, 17,1075.doi:10.1016/j.str.2009.06.012
August 13,2012;Revised:September 7,2012;Published on Web:September 7,2012.
Mechanism of 3D Domain Swapping for Mpro-C:Clues from Molecular Simulations
HUANG Yong-Qi1,2,3KANG Xue1,4XIA Bin1,4,5LIU Zhi-Rong1,2,3,*
(1College of Chemistry and Molecular Engineering,Peking University,Beijing 100871,P.R.China;2Beijing National Laboratory for Molecular Sciences,State Key Laboratory for Structural Chemistry of Unstable and Stable Species,Peking University, Beijing 100871,P.R.China; 3Center for Quantitative Biology,Peking University,Beijing 100871,P.R.China;4Beijing Nuclear Magnetic Resonance Center,Peking University,Beijing 100871,P.R.China; 5School of Life Sciences,Peking University,Beijing 100871,P.R.China)
SARS coronavirus main protease(Mpro)is a key enzyme involved in the extensive proteolytic processing of the virusʹpolyproteins.The crystal structure of Mproreveals that the enzyme exists in two differenthomo-dimericforms:a three-dimensional(3D)domain-swapped form;and a non-3D domain-swapped form.The isolated C-terminal domain(Mpro-C)also forms a 3D domain-swapped structure similar to the full-length protein.Unlike conventional 3D domain-swapped structures,in which the swapped regions are located on the surface,Mpro-C swaps a helix at the core of a folded domain.In this work,we used molecular dynamics simulations and 3D domain-swapping predictions to investigate how a highly buried core helix in the helix bundle structure of Mpro-C can be swapped.We found that both structure-and sequence-based methods failed to predict the location of the hinge loop in Mpro-C and Mpro.Extensive molecular dynamics simulations were performed to investigate the structural properties of the unfolded monomer and the 3D domain-swapped dimer of Mpro-C.We found that,although the swapped region was buried in the native state,it was exposed in the unfolded monomer.Our results suggest that the opening of the swapped region in the fully or partially unfolded state may promote interactions between monomers and the formation of domain-swapped dimers.
SARS coronavirus;Main protease;Molecular simulation;Domain swapping; Protein-protein interaction; Protein unfolding
10.3866/PKU.WHXB201209072
∗Corresponding author.Email:LiuZhiRong@pku.edu.cn;Tel:+86-10-62752541;Fax:+86-10-62759595.
The project was supported by the National Key Basic Research Program of China(973)(2009CB918500,2003CB514104)and National Natural Science Foundation of China(20973016,11021463,31170682).
国家重点基础研究发展计划项目(973)(2009CB918500,2003CB514104)和国家自然科学基金(20973016,11021463,31170682)资助
O641
猜你喜欢
经济与管理(2022年2期)2022-03-24
建材发展导向(2021年14期)2021-08-23
经济与管理(2021年2期)2021-03-15
艺术品鉴(2020年5期)2020-07-27
中国煤层气(2019年2期)2019-08-27
环境与可持续发展(2017年2期)2017-04-06
法语学习(2016年1期)2016-12-18
中国卫生标准管理(2015年16期)2016-01-20
现代检验医学杂志(2015年2期)2015-02-06
现代检验医学杂志(2015年1期)2015-02-06
