
Au12M(M=Na,Mg,Al,Si,P,S,Cl)团簇的结构、稳定性和电子性质
2012-12-21 06:33赵高峰王银亮孙建敏王渊旭
物理化学学报 2012年6期
赵高峰 王银亮 孙建敏 王渊旭
(河南大学计算材料科学研究所,河南开封475004)
Au12M(M=Na,Mg,Al,Si,P,S,Cl)团簇的结构、稳定性和电子性质
赵高峰*王银亮 孙建敏 王渊旭
(河南大学计算材料科学研究所,河南开封475004)
采用基于密度泛函理论的第一性原理方法系统地研究了Au12M(M=Na,Mg,Al,Si,P,S,Cl)团簇的结构、稳定性和电子性质.对团簇的平均结合能、镶嵌能、垂直离化势、最高占据分子轨道(HOMO)和最低未占据分子轨道(LUMO)的能级差、电荷布居分析、自然键轨道(NBO)进行了计算和讨论.对于Au12M(M=Na,Mg,Al)团簇,它们形成了内含M原子的最稳定的笼状结构.然而对于Au12M(M=Si,P,S,Cl)团簇,它们却形成了以M元素为顶点的稳定锥形结构.在这些团簇中发现Au12S团簇相对是最稳定的,这是由于Au12S团簇形成了稳定的满壳层的电子结构.自然电荷布居分析表明:对于所有的Au12M(M=Na,Mg,Al,Si,P,S,Cl)团簇电荷总是从Au原子转向M原子.自然键轨道和HOMO分析表明Au12M团簇中发生了Au原子的s-d轨道和M原子的p轨道间的杂化现象.
密度泛函理论;团簇;自然电荷布居分析;稳定性;自然键轨道分析
1 Introduction
During the past two decades,coinage metal clusters have been intensively studied by both experimental and theoretical methods.Clustering occurs due to the facile hybridization of core d-electrons with outer s-electrons.Gold clusters have been of particular interest.Recently,Bulusu et al.1reported evidence of hollow cages of pure metal atoms.A novel Au20tetrahedral structure identified by photoelectron spectroscopy correlates with relativistic density functional theory(DFT)calculations.2Fa and Dong3identified a stable tube-like Aun(n= 26-28)cluster with scalar,relativistic,all-electron DFT.Highly stable“golden fullerene”Au32andAu42clusters have been reported,4,5and core-shell structures have been verified by recent studies on Au34and Au58clusters.6-8The existence of these high-symmetry clusters is attributed to the manifestation of aurophilicity,which can further enhance strong gold-gold interactions.9In addition,relativistic-effect-enhanced s-d hybridization and s-electron delocalization may also reflect the preference for high-symmetry structures.10-12
Doping of gold clusters with impurity atoms is expected to open up new channels in which one can tailor properties by varying the nature of the dopant atom.13-15Since Pykko16and Li17et al.first reported the existence of highly stable Au12Wvia photoelectron spectroscopy,a considerable amount of experimental and theoretical work has been carried out on Au clusters doped with other impurity atoms.11,18-34Most of these studies have focused on Au12doped with transition-metal(TM)atoms.The high Ihor Ohsymmetry of the lowest-energy Au12TM clusters is attributed to the strong relativistic effect,aurophilic attraction,and 18-electron bonding to the 4s,5s,and 6s shells of the central heteroatom.16,35Furthermore,Au12TM clusters are more stable relative to icosahedral Au12and Au13cages,as verified by previous experimental17and theoretical11results.
It is thus clear that the ground-state geometries of Au12TM clusters are icosahedral or octahedral,the reason being that TM atoms possess outer s electron shells.Although a number of studies have focused on the geometric structures and electronic properties of Au12TM clusters,there have been relatively few studies on gold clusters doped with non-transition elements.24,25,36-42In this paper,we perform first-principles studies of single atom impurities with 3s and 3p electrons in Au12clusters.These impurity atoms come from the same row of the Periodic Table,thus their principle quantum numbers remain the same while having an increasing number of valence electrons.When these atoms are embedded in Au12clusters,however,there are clear differences in their lowest-energy Au12M structures.
2 Computational details
All computations were performed by DFT with the unrestricted B3LYP exchange-correlation potential43-48and the effective core potential standard LanL2DZ basis sets.49-51The standard LanL2DZ basis sets are effective in calculating noble metals because they reduce difficulties in two-electron integral calculations caused by the heavy atoms.
Calculations were performed with the Gaussian 03 program package.52For each stationary point of a cluster,the stability was examined by calculating the harmonic vibrational frequencies.If an imaginary frequency was found,a relaxation along the coordinates of the imaginary vibrational mode was carried out until a true local minimum was obtained.Therefore,all isomers for each cluster are guaranteed to be the local minimum. In addition,for the geometry optimization of each isomer,the spin multiplicity(SM)was at least 1,3,and 5 for even-electron clusters(Mg,Si,S,)and 2,4,and 6 for odd-electron clusters (Al,P,Cl).If the total energy decreases with increasing SM, we would use a higher spin state until the energy minimum was found.
In order to test the validity of the computational method,we performed calculations on Au2and AuAl dimers.As illustrated in Table 1,our results are in good agreement with previous experimental and theoretical data.25,53-59
3 Results and discussion
3.1 Structures of clusters
We examined a considerable number of low-lying isomers and determined the lowest-energy structures for Au12M(M= Na,Mg,Al,Si,P,Cl)clusters that are illustrated in Fig.1.For comparison,the icosahedral and octahedra cages for pure Au13clusters are also in Fig.1.In order to explain the structural features of these lowest-energy structures,we list the point group symmetry,the smallest bond length for Au-Au and Au-M, and the spin multiplicity in Table 2.
Previous studies indicate that the ground-state structures of Au12TM clusters have TM encapsulated in the center of Au12icosahedral or octahedral cages with high Ihor Ohsymmetry.11,16,17,26,27In our work,the lowest-energy structures of Au12M (M=Na,Mg)clusters are similar to the octahedral structures of Au12TM clusters.However,the other Au12M(M=Al,Si,P,S, Cl)structures differ from theAu12TM structures.
The ground state of the Au12Na cluster is an octahedral structure with the Na atom at its center,with D3dsymmetry,and a spin multiplicity of 2.The icosahedral structure also has the Na atom in the Au12cage center;however,its energy is 1.36 eVhigher than the ground state.In the octahedral structure,the shortest bond lengths of Au-Au and Au-Na are 0.284 and 0.291 nm,respectively,while the shortest bond lengths of Au-Au and Au-Na are 0.297 and 0.283 nm,respectively,for the icosahedral structure.When an Mg atom imbeds in the Au12cluster,it also forms an octahedral structure with the Mg in the center.However,the symmetry(Oh)of Au12Mg is higher than that(D3d)of Au12Na because all the Au—Au and Au—Mg bond lengths are the same(0.288 nm).The next higher energy isomer Au12Mg(b)in Fig.1 has S4point group symmetry with an energy very close to the ground-state structure(ΔE=0.59 eV). Recently,the geometric and electronic structures of clusters with a central 3d,4d,and 5d transition-metal atom encapsulated in an Au12cage have been investigated.11,26For encapsulated 3d and 4d transition-metals,the icosahedral clusters tend to be more stable than their octahedral isomers.But for 5d transitionmetals,the octahedral clusters tend to be more stable than their icosahedral isomers(except for Au12W).The octahedral structures of Au12Na and Au12Mg are more stable than their icosahedral isomers.Thus their ground state structures are similar to the clusters with a central 5d transition-metal(except for Au12W),but they differ from those with 3d,4d transition-metal impurities.In the case of Au12Al,the ground-state structure can be seen as a deformed octahedron with D2hsymmetry.Although the Al atom remains at the center,the outer Au12octahedral cage undergoes severe deformation.
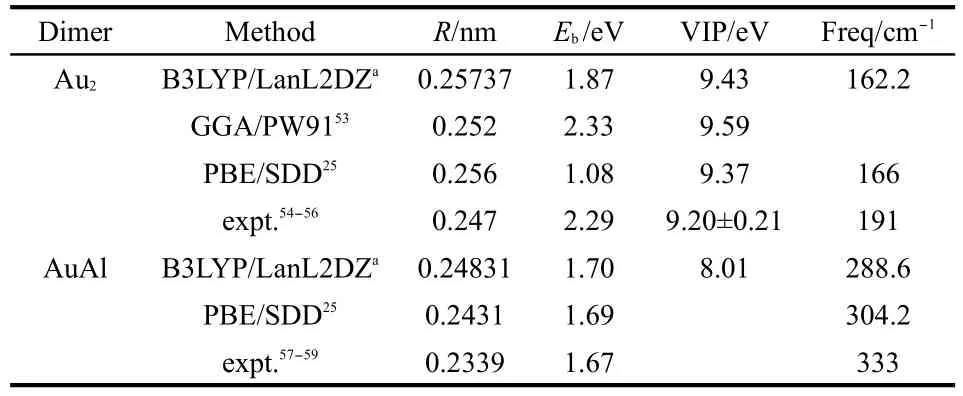
Table 1 Bond lengths(R),lowest harmonic vibrational frequencies(Freq),average binding energies(Eb),and vertical ionization potentials(VIPs)for the ground states ofAu2and AuAl dimers
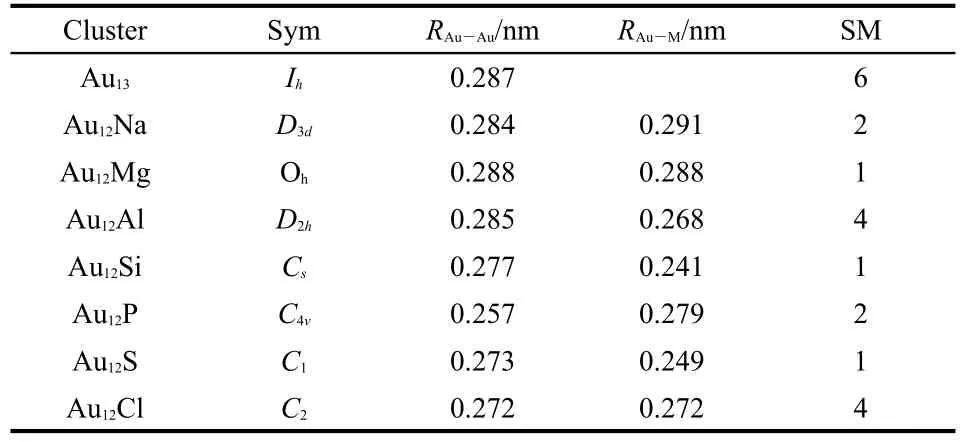
Table 2 Geometries of the lowest-energy isomers ofAu12M(M= Na,Mg,Al,Si,P,S,Cl)andAu13clusters
The first important change occurs in the lowest-energy structure of Au12Si,where the Si atom is now located at the top of a pyramid formed by the Au atoms.The pyramidal structure possesses Cssymmetry and a spin multiplicity of 1.The fact that the Si atom is not encapsulated in the Au12cage as for Au12Na and Au12Mg may be due to the bonding properties and the orbital hybridization between M and Au atoms.The octahedral Au12Si cluster has an energy that is 1.45 eV higher than the pyramidal isomer.The ground-state structure of Au12P is also a pyramid,however it has higher symmetry(C4v)compared to Au12Si.The shortest bond lengths of Au-P and Au-Au are 0.257 and 0.279 nm.As shown in Fig.1,the Au12P cluster is more compact than Au12Si,which may be attributed to different Au-Si andAu-P bondings.
The lowest-energy structure for Au12S is an irregular flat pyramid with low symmetry(C1),with the S atom at the bottom (Fig.1).It is thus more flat and extended than Au12P and Au12Si. It can be argued that the structure of Au12S results from electron delocalization over all the atoms.Surprisingly,a planar rhombic structure of Au12S is also observed,where the S atom occupies the center of the plane.However,its energy is 1.38 eV higher than the ground-state structure.Finally,we note that the Au12Cl cluster has a lowest-energy structure that is basketlike with the Cl atom at the apex.
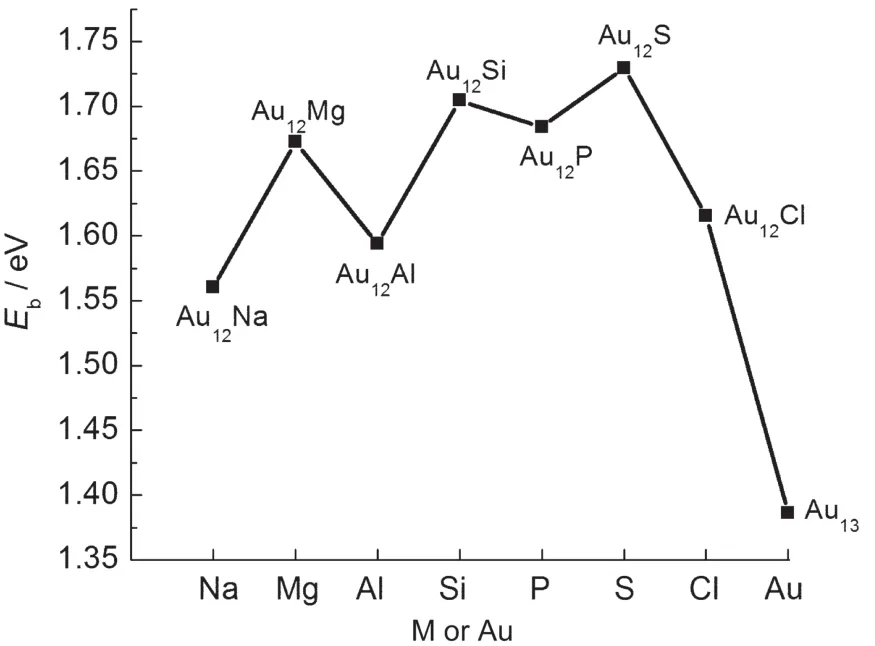
Fig.2 Average binding energies(Eb)of ground-stateAu12M (M=Na,Mg,Al,Si,P,S,Cl)andAu13clusters
3.2 Stabilities of clusters
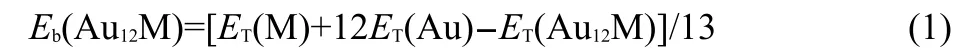
The average binding energy(Eb)of a given cluster is a measure of its thermodynamic stability,which is defined as the difference between the energy sum of all the free atoms constituting the cluster and the total energy of the cluster,as given by: where ET(M),ET(Au),and ET(Au12M)represent the total energies of the lowest-energy M,Au,and Au12M,respectively.As seen from Fig.2,the Ebfor the ground states of Au12M(M=Na, Mg,Al,Si,P,S,Cl)clusters are higher than that of the pure Au13cluster.The Au12S cluster,possessing the largest Eb,is also found to be the most stable under study.This is attributed to the closed-shell(18-electron shell-filling)rule,with one electron from eachAu atom and six electrons from the S atom.
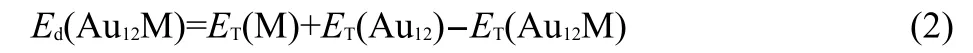
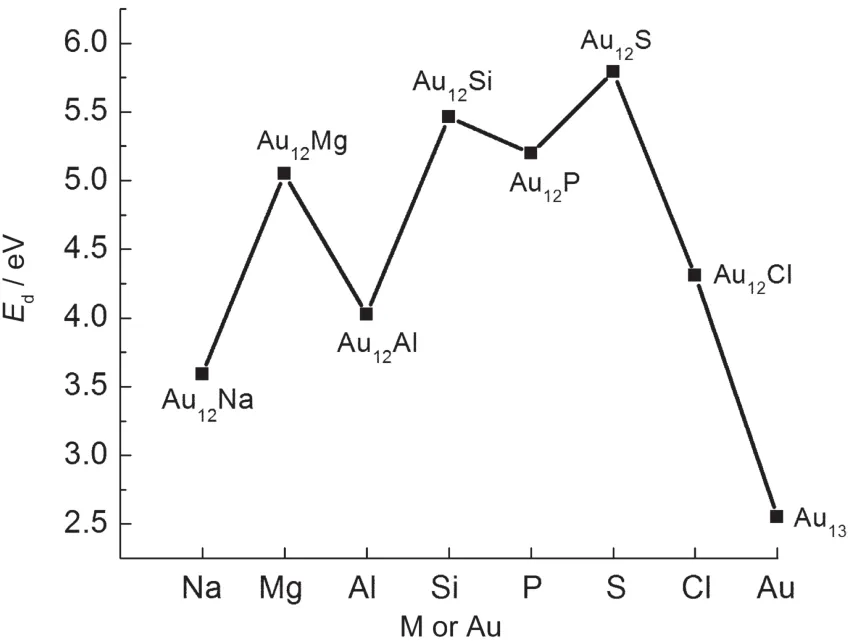
Fig.3 Embedding energies of ground-stateAu12M (M=Na,Mg,Al,Si,P,S,Cl)andAu13clusters
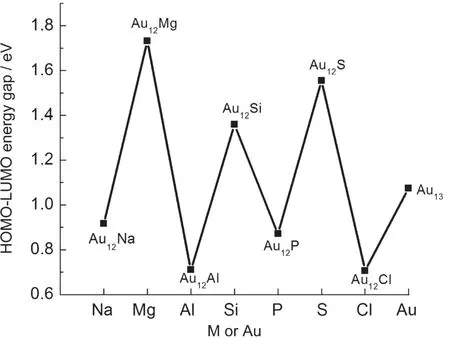
Fig.4 HOMO-LUMO energy gaps in ground-stateAu12M (M=Na,Mg,Al,Si,P,S,Cl)andAu13clusters
To further understand the stabilities of Au12M clusters,we will discuss the embedding energy(Ed)of the ground-state structure,which is defined as: where ET(M),ET(Au12),and ET(Au12M)represent the total energies of the lowest-energy M,Au12,and Au12M clusters,respectively.As shown in Fig.3,Au12S possesses the highest embedding energy among Au12M(M=Na,Mg,Al,Si,P,S,Cl)clusters.Hence,Au12S should be the most stable.
The energy gap between the highest occupied molecular orbital and the lowest unoccupied molecular orbital(HOMOLUMO)is a useful quantity when examining the chemical stability of clusters.A large energy gap correlates with a high barrier required to perturb the electronic structure.HOMOLUMO energy gaps for ground-state Au12M(M=Na,Mg,Al, Si,P,S,Cl)and Au13clusters are displayed in Fig.4.The largest energy gap(1.73 eV)is for Au12Mg,which indicates that it is the most chemically stable of these clusters.Meanwhile, Au12S has the second highest energy gap,and since it has the largest average binding energy among these clusters,it is both chemically and thermodynamically stable.

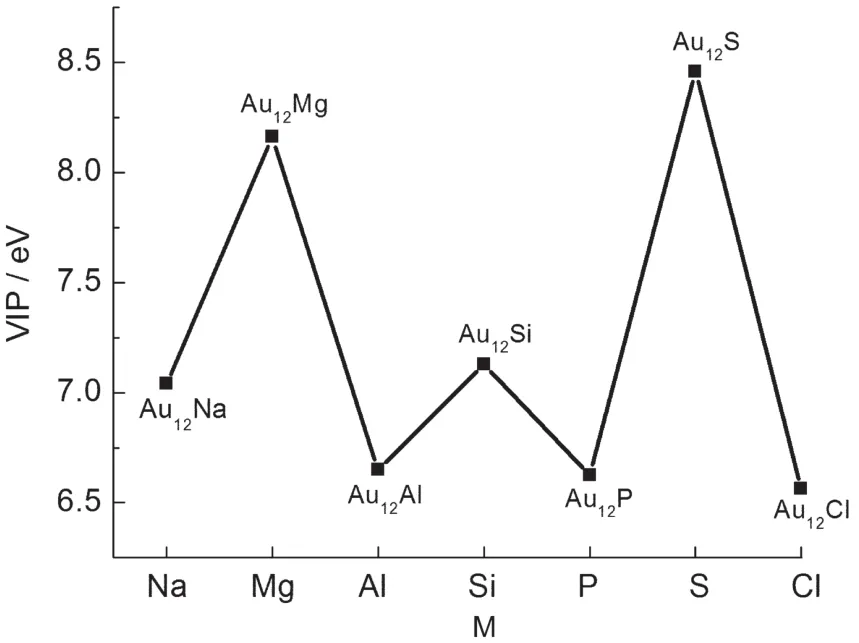
Fig.5 VIPof ground-stateAu12M(M=Na,Mg,Al,Si,P,S,Cl) clusters
The vertical ionization potential(VIP)is yet another parameter used to assess the chemical stability of small clusters,and is given by: where ET(Au12M+)is the total energy of the ionic clusters at the optimized neutral geometry.Large VIPs indicate high chemical stability.As shown in Fig.5,the VIPs of Au12Mg,Au12Si,and Au12S clusters are surprisingly higher than those for Au12Na, Au12Al,Au12P,and Au12Cl.This trend may be attributed to the number of electrons;recall that Au12Mg,Au12Si,and Au12S possess electrons in closed-shells,while the other four have electrons in open-shells.It indicates that Au12M(M=Mg,Si,S) clusters are chemically more stable than the other Au12M(M= Na,Al,P,Cl)clusters.Additionally,the VIP of Au12S is the largest in this series,which can be explained on the basis of its full closed-shells(18-electron rule).60
3.3 Electronic properties
Charge-transfer phenomena in the Au12M clusters can be obtained by natural population analysis.The atomic charges of the M atoms in the ground-state Au12M(M=Na,Mg,Al,Si,P, S,Cl)clusters are listed in Table 3,where we see that charges always transfer from the Au atoms to the electron-accepting M atoms.This clearly differs from that observed for Au5M and Au6M(M=Na,Mg,Al,Si,P,S,Cl)clusters.24,25Thus an important finding is that the direction of charge-transfer in M-doped gold clusters depends on cluster size.
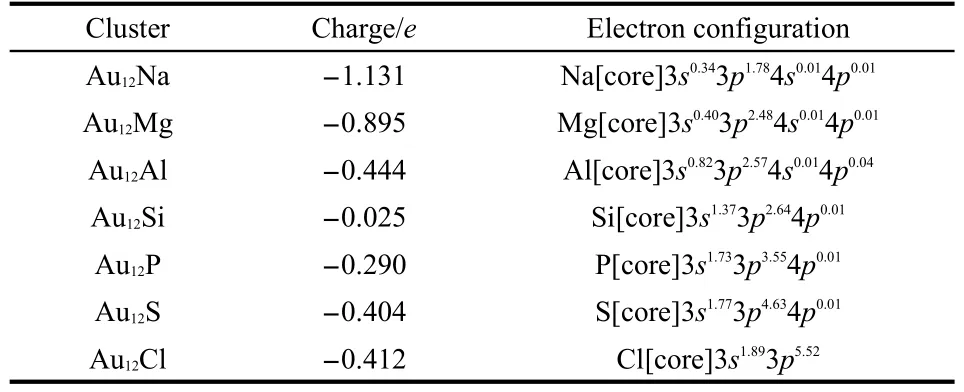
Table 3 Natural charge population and the electron configurations for M atoms inAu12M(M=Na,Mg,Al,Si,P,S,Cl) clusters through natural bond orbital(NBO)analysis
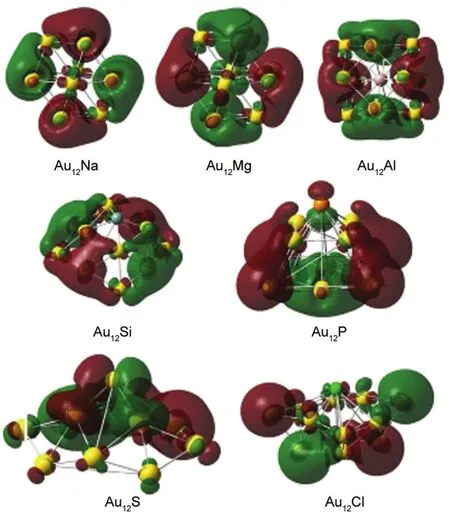
Fig.6 Spatial orientation of the highest occupied molecular orbitals of the ground-stateAu12M(M=Na,Mg,Al,Si,P,S,Cl) clusters
To examine hybridization between M(M=Na,Mg,Al,Si,P, S,Cl)atoms and Au in Au12M clusters,we present in Table 3 the natural electron configurations obtained from natural bond orbital(NBO)analysis.The valence electron configurations of the free atoms Na,Mg,Al,Si,P,S,and Cl are 3s1,3s2,3s23p1, 3s23p2,3s23p3,3s23p4,and 3s23p5,respectively.The NBO analysis in Table 3 reflects this s-p hybridization,where electrons transfer mainly from 3s to 3p orbitals in the M atoms.We also note that electrons transfer from 6s and 5d orbitals to 6p orbitals in the Au atoms,indicating sd-p hybridization.Since the 3p orbital gains more than the 3s orbital loses in the M atoms,it follows that the 6s and 5d orbitals in theAu atoms transfer electrons to the M 3p orbital.Thus hybridization does occur between the p orbital of the M atom and the s-d orbitals of the Au atoms.In order to further understand the chemical bonds in these systems,we plot in Fig.6 the spatial orientation of the HOMO energy levels for the Au12M clusters.The HOMOs show hybridization phenomena between p orbitals of the M atoms and the s-d orbitals of the Au atoms.These pictures are in good agreement with the NBO analysis.However,the hybridization of Au12M clusters differs from that in Au12TM,11which is attributed to the electronic properties of the dopant atoms.
4 Summary
We have carried out a first-principles investigation using DFT to systematically study the geometries and electronic properties of Au12M(M=Na,Mg,Al,Si,P,S,Cl)clusters.The Au12M(M=Na,Mg,Al)clusters form the lowest-energy cage structures with the M atom encapsulated in the center,while Au12M(M=Si,P,S,Cl)clusters form pyramids with the M atom at the apex.The lowest-energy geometries of Au12M(except Au12Na and Au12Mg clusters)are different from the high symmetry structure of 3d,4d,and 5d transition-metals in Au12TM clusters.This indicates that impurity atoms play a critical role in determining the structures and properties of Au12M clusters.The Au12S cluster,having full closed-shell orbitals,not only possesses a relatively high average binding energy and doping energy,but also a high VIP and HOMO-LUMO energy gap.Thus it is more stable than the other Au12M clusters.Finally,we note that an NBO analysis reveals that hybridization between the s-d orbitals in Au atoms and the p orbitals of the M impurities occurs inAu12M clusters.
(1) Bulusu,S.;Li,X.;Wang,L.S.;Zeng,X.C.Proc.Natl.Acad. Sci.U.S.A.2006,103,8326.
(2) Li,J.;Li,X.;Zhai,H.J.;Wang,L.S.Science 2003,299,864.
(3)Fa,W.;Dong,J.M.J.Chem.Phys.2006,124,114310.
(4)Johansson,M.P.;Sundholm,D.;Vaara,J.Angew.Chem.Int. Edit.2004,43,2678.
(5)Gao,Y.;Zeng,X.C.J.Am.Chem.Soc.2005,127,3698.
(6)Gu,X.;Bulusu,S.;Li,X.;Zeng,X.C.;Li,J.;Gong,X.G.; Wang,L.S.J.Phys.Chem.C 2007,111,8228.
(7)Huang,W.;Ji,M.;Dong,C.D.;Gu,X.;Wang,L.M.;Gong,X. G.;Wang,L.S.ACS Nano 2008,2,897.
(8)Dong,C.D.;Gong,X.G.J.Chem.Phys.2010,132,104301.
(9)Scherbaum,F.;Grohmann,A.;Huber,B.;Krueger,C.; Schmidbaur,H.Angew.Chem.1988,100,1602.
(10) Pyykko,P.Chem.Rev.1988,88,563.
(11) Wang,S.Y.;Yu,J.Z.;Mizuseki,H.;Sun,Q.;Wang,C.Y.; Kawazoe,Y.Phys.Rev.B 2004,70,165413.
(12)Hakkinen,H.;Moseler,M.;Kostko,O.;Morgner,N.; Hoffmann,M.A.;Issendorff,B.V.Phys.Rev.Lett 2004,93, 093401.
(13)Yu,Y.J.;Wang,H.Y.;Yang,C.L.;Chen,J.N.ActaPhys.-Chim. Sin.2011,27,808.[于永江,王华阳,杨传路,陈建农.物理化学学报,2011,27,808.]
(14) Qian,H.F.;Barry,E.;Zhu,Y.;Jin,R.C.Acta Phys.-Chim.Sin. 2011,27,513.
(15)Liang,W.H.;Wang,X.L.;Ding,X.C.;Chu,L.Z.;Deng,Z.C.; Fu,G.S.;Wang,Y.L.Acta Phys.-Chim.Sin.2011,27,1615. [梁伟华,王秀丽,丁学成,禇立志,邓泽超,傅广生,王英龙.物理化学学报,2011,27,1615.]
(16)Pykko,P.;Runeberg,N.Angew.Chem.2002,41,2174.
(17)Li,X.;Kiran,B.;Li,H.;Zhai,H.J.;Wang,L.S.Angew.Chem. Int.Edit.2002,41,4786.
(18)Chen,M.X.;Yan,X.H.J.Chem.Phys.2008,128,174305.
(19) Heinebrodt,M.;Malinowski,N.;Tast,F.;Branz,W.;Billas,I. M.L.;Martin,T.P.J.Chem.Phys.1996,110,9915.
(20)Huang,W.;Wang,L.S.Phys.Rev.Lett.2009,102,153401.
(21)Wang,L.M.;Pal,R.;Huang,W.;Zeng,X.C.;Wang,L.S. J.Chem.Phys.2010,132,114306.
(22)Ferrighi,L.;Hammer,B.;Madsen,G.K.H.J.Am.Chem.Soc. 2009,131,10605.
(23)Zhang,M.;He,L.M.;Zhao,L.X.;Feng,X.J.;Luo,Y.H. J.Phys.Chem.C 2009,113,6491.
(24)Majumder,C.K.;Kandalam,A.K.;Jena,P.Phys.Rev.B 2006, 74,205437.
(25)Zhang,M.;Chen,S.;Deng,Q.M.;He,L.M.;Zhao,L.N.;Luo, Y.H.Eur.Phys.J.D 2010,58,117.
(26)Long,J.;Qiu,Y.X.;Chen,X.Y.;Wang,S.G.J.Phys.Chem.C 2008,112,12646.
(27) Zhai,H.J.;Li,J.;Wang,L.S.J.Chem.Phys.2004,121,8369.
(28)Gao,Y.;Bulusu,S.;Zeng,X.C.ChemPhysChem 2006,7,2275. (29) Li,X.;Kiran,B.;Cui,L.F.;Wang,L.S.Phys.Rev.Lett.2005, 95,253401.
(30)Yang,A.P.;Fa,W.;Dong,J.M.J.Phys.Chem.A 2010,114, 4031.
(31)Sun,Q.;Wang,Q.;Jena,P.;Kawazoe,Y.ACS Nano 2008,2, 341.
(32)Wang,L.M.;Bai,J.;Lechtken,A.;Huang,W.;Schooss,D.; Kappes,M.M.;Zeng,X.C.;Wang,L.S.Phys.Rev.B 2009,79, 033413.
(33) Neukermans,S.;Janssens,E.;Tanaka,H.;Silverans,R.E.; Lievens,P.Phys.Rev.Lett.2003,90,033401.
(34)Walter,M.;Hakkinen,H.Phys.Chem.Chem.Phys.2006,8, 5407.
(35) Autschbach,J.;Hess,B.A.;Johansson,M.P.;Neugebauer,J.; Patzschke,M.;Pyykko,P.;Reiher,M.;Sundholm,D.Phys. Chem.Chem.Phys.2004,6,11.
(36)Zhao,L.X.;Cao,T.T.;Feng,X.J.;Liang,X.;Lei,Y.M.;Luo, Y.H.J.Mol.Struct.-Theochem 2009,895,92.
(37) Graciela,B.P.;Ignacio,L.G.J.Mol.Struct.-Theochem 2002, 619,79.
(38) Banerjee,A.;Ghanty,T.K.;Chakrabarti,A.;Kamal,C.J.Phys. Chem.C 2012,116,193.
(39)Chen,D.D.;Kuang,X.Y.;Zhao,Y.R.;Shao,P.;Li,Y.F.Chin. Phys.B 2011,20,063601.
(40)Li,Y.F.;Kuang,X.Y.;Wang,S.J.J.Phys.Chem.A 2010,114, 11691.
(41) Jayasekharan,T.;Ghanty,T.K.J.Phys.Chem.C 2010,114, 8787.
(42) Zhao,L.X.;Feng,X.J.;Cao,T.T.;Liang,X.;Luo,Y.H.Chin. Phys.B 2009,18,2709.
(43) Becke,A.D.J.Chem.Phys.1986,84,4524.
(44) Becke,A.D.J.Chem.Phys.1988,88,2547.
(45) Becke,A.D.J.Chem.Phys.1988,88,1053.
(46)Lee,C.;Yang,W.;Parr,R.G.Phys.Rev.B 1988,37,785.
(47)Becke,A.D.J.Chem.Phys.1993,98,5468.
(48)Kohn,W.;Sham,L.J.Phys.Rev.A 1965,140,1133.
(49)Hay,P.J.;Wadt,W.R.J.Chem.Phys.1985,82,270.
(50)Hay,P.J.;Wadt,W.R.J.Chem.Phys.1985,82,299.
(51)Wadt,W.R.;Hay,P.J.J.Chem.Phys.1985,82,284.
(52) Frisch,M.J.;Trucks,G.W.;Schlegel,H.B.;et al.Gaussian 03, Revision B.03;Gaussian Inc.:Pittsburgh,PA,2003.
(53) Zhao,G.F.;Zeng,Z.J.Chem.Phys.2006,125,014303.
(54)Morse,M.D.Chem.Rev.1986,86,1049.
(55)Negishi,Y.;Nakamura,Y.;Nakajima,A.;Kaya,K.J.Chem. Phys.2001,115,3657.
(56) Simard,B.;Hackett,P.A.J.Mol.Spectrosc.1990,142,310.
(57)Gingerich,K.A.;Blue,G.D.J.Chem.Phys.1973,59,185.
(58) Ho,J.;Ervin,K.;Lineberger,W.J.Chem.Phys.1990,93,6987.
(59) Taylor,K.;Pettitte-Hall,C.;Cheshnovsky,O.;Smalley,R. J.Chem.Phys.1992,96,3319.
(60)Tomlman,C.A.Chem.Soc.Rev.1972,1,337.
February 14,2012;Revised:April 5,2012;Published on Web:April 6,2012.
Geometries,Stabilities and Electronic Properties of Au12M (M=Na,Mg,Al,Si,P,S,Cl)Clusters
ZHAO Gao-Feng*WANG Yin-Liang SUN Jian-Min WANG Yuan-Xu
(Institute of Computational Materials Science,Henan University,Kaifeng 475004,Henan Province,P.R.China)
The geometries,stabilities,and electronic properties of Au12M(M=Na,Mg,Al,Si,P,S,Cl) clusters were systematically investigated by using first-principlescalculationsbased on density functional theory(DFT).For each cluster,the average binding energy,the embedding energy,the vertical ionization potential,the energy gap between the highest occupied molecular orbital(HOMO)and the lowest unoccupied molecular orbital(LUMO),the natural charge population analysis,and the natural bond orbital analysis(NBO)were calculated.The lowest-energy structures of Au12M(M=Na,Mg,Al) clusters are cages with M encapsulated in the center,while structures of Au12M(M=Si,P,S,Cl)clusters are pyramidal with M at the apex.The Au12S cluster,having the full closed-shells,is the most stable. Furthermore,from the natural population analysis,it follows that charges transfer from Au to M in all the clusters.The NBO and HOMO analyses reveal that hybridization occurs between the Au s-d orbitals and the M p orbitals.
Density functional theory;Cluster;Natural charge population analysis;Stability; Natural bond orbital analysis
10.3866/PKU.WHXB201204063
∗Corresponding author.Email:zgf@henu.edu.cn;Tel:+86-378-3881602.
The project was supported by the National Natural Science Foundation of China(10804027,11011140321)and Natural Science Foundation of Education Department of Henan Province,China(2011A140003).
国家自然科学基金(10804027,11011140321)和河南省教育厅自然科学基金(2011A140003)资助项目
O641
猜你喜欢
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
中学生数理化·中考版(2021年10期)2021-11-22
中学生数理化(高中版.高考理化)(2020年10期)2020-10-27
空间科学学报(2020年6期)2020-07-21
空间科学学报(2020年6期)2020-01-08
环球时报(2019-12-05)2019-12-05
新高考·高一物理(2015年6期)2015-09-28
新高考·高一物理(2015年6期)2015-09-28
