
ERK1/2信号通路的活化参与人脐静脉内皮细胞向间充质转分化的过程*
2013-12-23 05:15邓元俊裴广畅许楚瓯常晓燕
华中科技大学学报(医学版) 2013年5期
刘 萍, 邓元俊, 裴广畅, 许楚瓯, 常晓燕, 曾 锐, 姚 颖, 徐 钢, 韩 敏
华中科技大学同济医学院附属同济医院肾内科,武汉 430030
肾间质纤维化是各种肾脏疾病进行性发展至终末期肾病的最后共同途径,其基本病理变化为肾间质肌纤维母细胞产生的细胞外基质沉积。肾间质肌成纤维细胞有多种可能的来源,包括肾间质原位成纤维细胞、周细胞的增殖或活化,肾小管上皮细胞向间充质转分化,毛细血管内皮细胞向间充质转分化(endothelial to mesenchymal transition,EndoMT),骨髓来源的成纤维细胞的迁移[1-2]。Zeisberg 等[3]发现在单侧输尿管梗阻性肾病,链脲佐菌素诱导的糖尿病肾病和Alport肾病3种小鼠模型中30%~50%的肌成纤维细胞来源于内皮细胞,提示EndoMT 是肾间质纤维化的重要机制之一。Li等[4]也证实在链脲佐菌素诱导的糖尿病肾病中存在EndoMT,参与肾间质纤维化的早期进展。一方面,EndoMT 是肌成纤维细胞的重要来源之一;另一方面,内皮细胞转分化后,管周毛细血管的屏障功能受损,毛细血管的通透性增加,促进炎性细胞渗出,并导致管周毛细血管网的匮乏及肾间质局部缺氧[5],加重了肾间质局部的炎症和氧化应激反应,形成炎症-氧化应激-缺氧的微环境,二者互为因果,相互作用,促进肾间质纤维化的进行性发展[6]。由此可见,EndoMT 是肾间质纤维化进展的重要机制之一。
与肾小管上皮细胞转分化相似,转化生长因子β(transforming growth factor,TGF-β)可诱导EndoMT 的发生。Medici等[7]研究证实TGF-β可诱导小鼠胚胎干细胞来源的内皮细胞转分化以及Snail的表达,且Snail的表达上调与TGF-β2诱导的Smad,MEK/ERK,PI3K 以及p38 MAPK 的活化有关。Chen等[8]研究也表明在系统性硬化的真皮层成纤维细胞中检测到TGF-β信号通路的活化和ERK1/2 的磷酸化。越来越多的研究也证明MAPK 信号通路在慢性肾脏病的病理进程中起着重要作用[9-10]。然而,MAPK 信号通路的激活是否也参与EndoMT 的发生迄今尚无报道。本研究旨在明确MAPK 信号通路在TGF-β1 诱导的EndoMT 过程的可能作用。
1 材料和方法
1.1 材料
人脐静脉内皮细胞系(human umbilical vein endothelial cells,HUVECs)购自南京凯基生物有限公司。DMEM/1640培养液、胰蛋白酶购自南京凯基公司。胎牛血清购自Hyclone 公司。重组人TGF-β1购自R & D 公司。p38 MAPK 抑制剂SB203580、JNK 抑制剂SP600125均购自Sigma公司,ERK1/2特异性抑制剂U0126购自Cell Signaling公司。多克隆兔抗α-SMA 抗体购自Abcam 公司。单克隆小鼠抗VE-cadherin的抗体购自BD 公司,单克隆小鼠抗GAPDH 抗体购自Epitomic公司。CY3和FITC标记的荧光二抗购自于Jackson公司,DAPI购自罗氏公司。
1.2 细胞培养和干预
HUVECs培养于含10%胎牛血清的DMEM 1640培养液中,置于37℃、5%CO2培养箱中。细胞生长至80%~90%融合状态,予0.25%胰蛋白酶消化传代至6 孔板中。当细胞密度达到60%~80%时,无血清化24h后,加入TGF-β1(5ng/mL)刺激0、24、48、72h,并行相关检测。抑制剂干预实验分组如下:对照组,TGF-β1 模型组,TGF-β1 +DMSO组,TGF-β1+抑制剂组。TGF-β1模型组:予5ng/mL TGF-β1刺激HUVECs 72h;TGF-β1+DMSO组:予1μL/mL DMSO 干预HUVECs 30min后,再予5ng/mL TGF-β1刺激细胞72h;TGF-β1+抑制剂组:予p38 MAPK 特异性抑制剂SB203580(5 mmol/L)或JNK 特异性抑制剂SP600125 (5 mmol/L)或ERK1/2 特异性抑制剂U0126 (10 mmol/L)干预HUVECs 30 min 后,再加入5ng/mL TGF-β1刺激细胞72h。
1.3 Western blot法检测细胞蛋白表达
按1.2的方法干预细胞后,加入RIPA 裂解液裂解细胞,BCA 法进行蛋白定量。10%SDS聚丙烯酰胺凝胶电泳分离,电印迹转移法转至PVDF 膜上,封闭1h,再分别与特异性一抗anti-VE-cadherin(1∶300)、anti-α-SMA(1∶1 000)、anti-GAPDH(1∶4 000),4℃,孵育过夜,TBST 漂洗3次,HRP标记的二抗,37℃,孵育1h,TBST 清洗3次后,ECL显影,Quantity One图像分析软件测定条带的灰度值,目的蛋白的相对表达量以目的条带与对照蛋白(GAPDH)的相对灰度值表达。每组实验均重复3次。
1.4 免疫荧光法检测细胞蛋白表达
细胞以1×105/mL 传代至底部放有玻片的12孔板,分组为对照组、TGF-β1(5ng/mL)组,72h后经PBS 漂洗,予冰甲醇∶丙酮(1∶1)固定细胞,-20℃静置15min。室温下,0.5%Triton破膜10 min。5%BSA 室温封闭30min。分别予anti-VEcadherin(1∶25)、anti-α-SMA(1∶100),4℃,孵育过夜。PBS漂洗后,加入二抗,37℃,避光孵育1h。DAPI(1∶2 000),室温,避光孵育10 min。封片后,避光保存,镜检。
1.5 统计学分析
采用统计软件IBM SPSS statistics 19 进行统计学分析,计量数据以均数±标准差表示,各组组间比较采用t检验或单因素方差分析,以P<0.05为差异有统计学意义。
2 结果
2.1 TGF-β1可诱导HUVECs向间充质细胞转分化
分别用TGF-β1(5ng/mL)刺激HUVECs 0、24、48、72h。倒置显微镜(×200)观察细胞形态发现:随着刺激时间的延长,内皮细胞逐渐失去其铺路石样外观,呈现出成纤维细胞的梭长形改变(图1A)。TGF-β1(5ng/mL)刺激HUVECs 72h 后,收获细胞,应用免疫荧光法和Western blot法检测内皮细胞特异性标志蛋白VE-cadherin和间充质标志性蛋白α-SMA 的表达,结果显示:与0h 比较,TGF-β1刺激HUVECs 72h后,细胞膜几乎无内皮细胞标志性蛋白VE-cadherin的表达,而内皮细胞胞质内间充质标志性蛋白α-SMA 表达上调(图1B);Western blot结果也证实TGF-β1 刺激HUVECs 72h后,内皮细胞VE-cadherin蛋白表达下调(P<0.01),α-SMA 表达显著性增多(P<0.05)(图1C、D)。
2.2 p38 MAPK 的活化对EndoMT的影响
p38MAPK 特异性抑制剂SB203580干预HUVECs后,内皮细胞形态呈长梭形(图2A),与TGFβ1模型组和TGF-β1+DMSO 组比较,细胞形态无明显差异。VE-cadherin 和α-SMA 蛋白表达差异均无统计学意义(图2B);与对照组比较,VE-cadherin表达显著性下调(P<0.05),α-SMA 表达显著性上调(P<0.05)(图2C)。
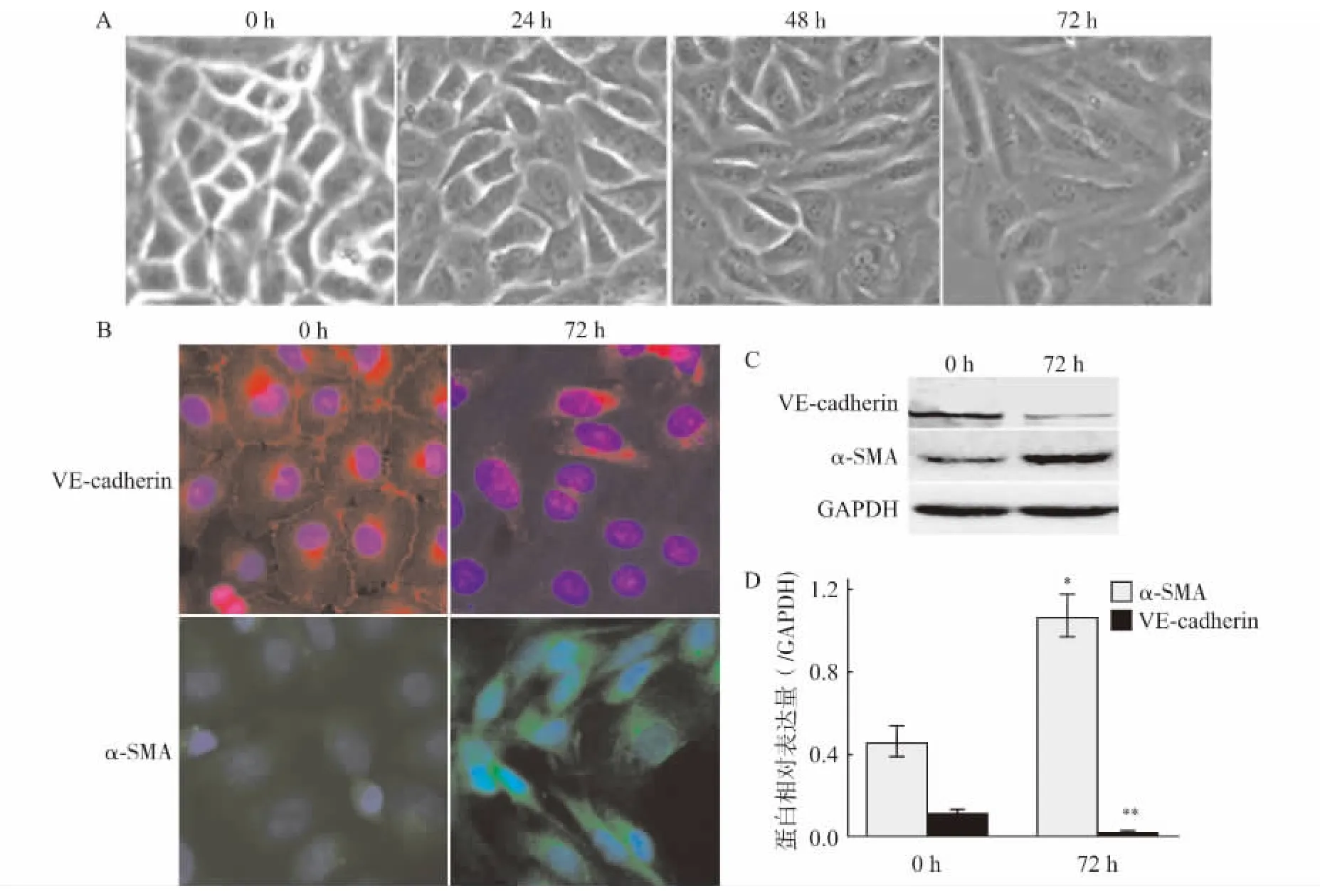
图1 TGF-β1刺激HUVECs向间充质转分化Fig.1 TGF-β1-induced endothelial-to-mesenchymal transition in HUVECs

图2 p38 MAPK 特异性抑制剂SB203580干预HUVECs后细胞形态和VE-cadherin、α-SMA 蛋白水平的改变Fig.2 The changes of cell morphology and expression of VE-cadherin andα-SMA in HUVECs treated with the p38 MAPK specific inhibitor SB203580
2.3 JNK 的活化对EndoMT的影响
JNK 特异性抑制剂SP600125 干预HUVECs后,内皮细胞形态呈长梭形(图3A),与TGF-β1模型组和TGF-β1+DMSO 组比较,细胞形态无明显差异。VE-cadherin和α-SMA 蛋白表达差异均无统计学意义(图3B);与对照组比较,VE-cadherin表达显著性下调(P<0.05),α-SMA 表达显著性上调(P<0.05)(图3C)。

图3 JNK 特异性抑制剂SP600125干预HUVECs后细胞形态和VE-cadherin、α-SMA 蛋白水平的改变Fig.3 The changes of cell morphology and expression of VE-cadherin andα-SMA in HUVECs treated with the JNK specific inhibitor SP600125
2.4 ERK1/2的活化对EndoMT的影响
ERK1/2特异性抑制剂U0126干预HUVECs后,内皮细胞形态呈铺路石状(图4A),与对照组比较,VE-cadherin和α-SMA 的表达差异无统计学意义(图4B);与TGF-β1模型组和TGF-β1+DMSO 组比较,VE-cadherin 表达显著上升(P <0.05),α-SMA 表达显著下降(P<0.05)(图4C)。

图4 ERK1/2特异性抑制剂U0126干预HUVECs后细胞形态和VE-cadherin、α-SMA 蛋白水平的改变Fig.4 The changes of cell morphology and expression of VE-cadherin andα-SMA in HUVECs treated with the ERK1/2specific inhibitor U0126
3 讨论
肾间质纤维化的本质被认为是一种“疤痕化”的修复过程。慢性炎症的持续存在刺激肌成纤维细胞的增殖活化,产生大量的细胞外基质,沉积于肾间质,最终导致肾单位的破坏和肾脏功能的下降[1]。肌成纤维细胞的来源一直备受研究者关注。有研究认为,肾小管管周毛细血管内皮细胞可能是肌成纤维细胞的重要来源之一[3],尤其在以血管病变为主的糖尿病肾病中,内皮细胞向间充质转分化可能是肌成纤维细胞的重要来源,参与肾间质纤维化的进行性发展[4]。然而,目前对于调控内皮细胞转分化的信号通路和转录机制的研究甚少,阐明肾小管管周毛细血管EndoMT 的信号调控途径将为探寻肾间质纤维化治疗的新靶点提供实验依据。
TGF-β1是肾间质纤维化进展的关键调控因子[7,11-12]。与肾小管上皮细胞转分化过程相似,TGF-β1也可诱导内皮细胞向间充质转分化。既往的研究表明,EndoMT 参与胚胎心脏瓣膜发育,而在此过程中检测到TGF-β1信号通路的活化[13]。本研究应用TGF-β1诱导人脐静脉内皮细胞向间充质细胞转分化,结果显示:TGF-β1(5ng/mL)刺激人脐静脉内皮细胞72h,不仅可使其形态发生变化而且内皮细胞标志性的粘附连接蛋白VE-cadherin表达下调,α-SMA 蛋白其表达上调,与0h 相比均存在统计学差异;细胞免疫荧光结果进一步证实这一现象。提示TGF-β1诱导HUVECs向间充质转分化的细胞模型制作成功,可用于EndoMT 的相关细胞研究。
大量的研究表明,MAPK 信号通路参与肾小管上皮细胞转分化的过程。本实验室既往的研究也表明:信号素蛋白Semaphorin 4C通过p38 MAPK 途径参与调控肾小管上皮细胞向间充质转分化[14];Erbin作为肾小管上皮细胞向间充质转分化的负性调节剂,主要通过ERK 1/2信号途径发挥作用[15]。在EndoMT 过程中,本研究结果显示:ERK1/2 信号通路参与调节TGF-β1诱导的HUVECs转分化。ERK1/2特异性抑制剂U0126可以显著减轻TGFβ1诱导的α-SMA 蛋白的表达上调,维持VE-cadherin在内皮细胞的表达;而抑制JNK 和p38 MAPK信号通路的活化对TGF-β1诱导的HUVECs转分化无显著影响。Medici等[16]的研究表明:TGF-β2刺激的EndoMT 过程,主要通过Smads信号通路和非Smads信号通路(包括MEK/ERK,PI3K 和p38 MAPK 信号通路)实现。IL-1β和TGF-β2协同诱导EndoMT 的过程中,Maleszewska等[17]的研究提示转录因子NF-κB是必不可少的。Li等[18]关于大鼠肺间质内皮细胞转分化研究则提示:c-Abl激酶和PKCδ是TGF-β1诱导的EndoMT 的关键调节因子。由此可见,在不同的组织中EndoMT 的信号调控通路可能不尽相同,Smads 通路、MAPK 通路(如ERK1/2通路)等多种通路均参与其中[19-20]。
尽管本研究已初步证实ERK1/2 信号通路是内皮细胞转分化的关键调节因子之一,但是本研究仅应用化学性信号通路的特异性抑制剂进行干预,存在一定的局限性,需进一步应用siRNA 和质粒等生物学技术进一步验证该信号通路对EndoMT 发生的影响,另外还需要在体内实验进行验证。另外,本实验尚未检测TGF-β/Smads信号通路的活化及转录因子的表达情况,Smads 信号通路是否与ERK1/2信号途径协同,参与EndoMT 的发生,还需要在后续的研究中进一步论证。
[1] Grgic I,Duffield J S,Humphreys B D.The origin of interstitial myofibroblasts in chronic kidney disease[J].Pediatric Nephrology,2011,27(2):183-193.
[2] Wiwanitkit V.Fibrosis and evidence for epithelial-mesenchymal transition in the kidneys of patients with staghorn calculi[J].BJU Int,2011,108(8):1336-1345.
[3] Zeisberg E M,Potenta S E,Sugimoto H.et al.Fibroblasts in kidney fibrosis emerge via endothelial-to-mesenchymal transition[J].J Am Soc Nephrol,2008,19(12):2282-2287.
[4] Li J,Qu X,Bertram J F.Endothelial-myofibroblast transition contributes to the early development of diabetic renal interstitial fibrosis in streptozotocin-induced diabetic mice[J].Am J Pathol,2009,175(4):1380-1388.
[5] Basile D P,Friedrich J L,Spahic J,et al.Impaired endothelial proliferation and mesenchymal transition contribute to vascular rare faction following acute kidney injury[J].Am J Physiol Renal Physiol,2011,300(3):721-733.
[6] López-Novoa J M,Nieto M A.Inflammation and EMT:an alliance towards organ fibrosis and cancer progression[J].Mol Med,2009,1(6):303-314.
[7] Medici D,Potenta S,Kalluri R.Transforming growth factorβ2promotes Snail-mediated endothelial-mesenchymal transitionthrough convergence of Smad-dependent and Smad-independentsignaling[J].Biochem J,2011,437(3):515-520
[8] Chen Y,Leask A,Abraham D J,et al.Thrombospondin 1is a key mediator of tranforming growth factorβ-mediated cell contractility in systemic sclerosis via a mitogen-activated protein kinase kinase(MEK)/extracellular signal-regulated kinase(ERK)-dependent mechanism[J].Fibrogenesis Tissue Repair,2011,4(1):9.
[9] Wang W,Koka V,Lan H Y.Transforming growth factor-beta and Smad signalling in kidney diseases[J].Nephrology(Carlton),2005,10(1):48-56.
[10] Eddy A A,Neilson E G.Chronic kidney disease progression[J].J Am Soc Nephrol,2006,17(11):2964-2966.
[11] Rieder F,Kessler S P,West G A,et al.Inflammation-induced endothelial-to-mesenchymal transition:a novel mechanism of intestinal fibrosis[J].Am J Pathol,2011,179(5):2660-2673.
[12] Varga J,Abraham D.Systemic sclerosis:aprototypic multisystem fibrotic disorder[J].J Clin Invest,2007,117(3):557-567.
[13] Boor P,Ostendorf T,Floege J.Renal fibrosis:novel insights into mechanisms and therapeutic targets[J].Nat Rev Nephrol,2010,6(11):643-656.
[14] Zeng R,Han M,Luo Y,et al.Role of Sema4Cin TGF-β1-induced mitogen-activated protein kinaseactivation and epithelial-mesenchymal transition in renal tubularepithelial cells[J].Nephrol Dial Transplant,2010,26(4):1149-1156.
[15] Zhou Q,Zeng R,Xu C,et al.Erbin inhibits TGF-β1-induced EMT in renal tubular epithelial cells through an ERK-dependent pathway[J].J Mol Med,2011,90(4):563-574.
[16] Medici D,Shore E M,Lounev V Y,et al.Conversion of vascular endothelial cells into multipotent stem-like cells[J].Nat Med,2010,16(12):1400-1406.
[17] Maleszewska M,Moonen J R,Huijkman N,et al.IL-1βand TGF-β2 synergistically induce endothelial to mesenchymal transition in an NFκB-dependent manner[J].Immunobiology,2013,218(4):443-454.
[18] Li Z,Jimenez S A.Protein kinase Cδand c-Abl kinase are required for transforming growth factorβinduction of endothelial-mesenchymal transition in vitro[J].Arthritis Rheum,2011,63(8):2473-2483.
[19] 李伟,邵乐南,张小燕,等.ERK1/2通路与舌鳞癌细胞Cal-27生物学行为的相关性[J].华中科技大学学报:医学版,2012,41(6):650-655.
[20] 肖玉霞,邵乐南,黄梅靖,等.PTPIP51、p-Raf-1及ERK1/2在口腔鳞状细胞癌中的表达[J].华中科技大学学报:医学版,2011,40(4):417-422.
猜你喜欢
昆明医科大学学报(2021年8期)2021-08-13
昆明医科大学学报(2021年3期)2021-07-22
昆明医科大学学报(2021年5期)2021-07-22
云南医药(2021年3期)2021-07-21
现代临床医学(2021年2期)2021-03-29
世界科学技术-中医药现代化(2021年10期)2021-03-02
中成药(2018年5期)2018-06-06
中国测试(2016年9期)2016-08-13
中国现代医学杂志(2015年26期)2015-12-23
中国现代医学杂志(2015年26期)2015-12-23
