
基于隐马尔可夫模型对原核生物编码序列的识别*
2015-03-09 11:13山西医科大学卫生统计教研室030001曹红艳张岩波
中国卫生统计 2015年2期
山西医科大学卫生统计教研室(030001) 曹红艳 马 靖 李 治 张岩波
基于隐马尔可夫模型对原核生物编码序列的识别*
山西医科大学卫生统计教研室(030001) 曹红艳 马 靖 李 治 张岩波△
目的探讨隐马尔可夫模型在大肠杆菌编码序列识别中的应用,为生物信息挖掘、致病位点研究提供方法参考。方法对大肠杆菌训练集数据进行训练建模,并对测试序列进行识别,用特异度、灵敏度以及精确度三个指标进行评价。结果利用本试验的方法识别编码序列的灵敏度为73.33%,特异度为67.78%,精确度为70.56%。结论隐马尔可夫模型能很好地模拟离散状态间的转换,适用于识别有状态转移、线性序列的数据。
隐马尔可夫模型 编码区序列识别 大肠杆菌
随着2003年人类基因组测序的完成,快速、准确的基因注释对进一步识别基因,解释生命的起源和进化等具有重要的意义[1]。基因注释包括识别出基因序列中的启动子、编码区、调控区等区域以及其他一些未被发现的功能片段,其关键问题是找出基因组中所有的基因,即基因识别过程[2]。其中,编码区域识别,即识别DNA序列中的编码蛋白质部分的序列,显得尤为重要。基于实验研究的基因识别耗时长、速度慢,远滞后于基因测序的速度,因此,需要寻求机器学习的方法用于基因识别[1]。
隐马尔可夫模型(hidden markov model,HMM)是当前机器学习的研究热点[3],由于HMM结构和基因结构非常吻合,且HMM基于坚实的数学理论体系,能保证精确的分析[4],另外,HMM的计算处理时间远小于伪支持向量机,对于海量的生物信息数据分析有很大的优势,因此,HMM广泛应用于生物信息领域[3]。本文采用HMM对原核生物编码序列进行识别,以期为生物信息挖掘、致病位点研究提供方法参考。
原理与方法
1.隐马尔可夫模型定义
隐马尔可夫模型是由马尔可夫链发展而来的一种随机模型,由两个随机过程{xk,yk,k=1,2,3,…}组成,其中{xk}是由离散隐状态组成的状态序列,称为路径或状态链,描述状态之间的转换;{yk}是由可观察字符组成的观察序列,称之为观测链,用来描述状态与观察值的对应关系。对编码序列进行识别时,序列的状态集合为{编码区Y,非编码区N},而序列是由四个碱基组成,故观察集合为{A,T,C,G}。作为观察者,不能直接看到状态,而是通过一个随机过程去感知状态的存在及其特性,因而称为“隐”马尔可夫模型[5]。图1表示了DNA序列识别的隐马尔可夫模型内部关系。
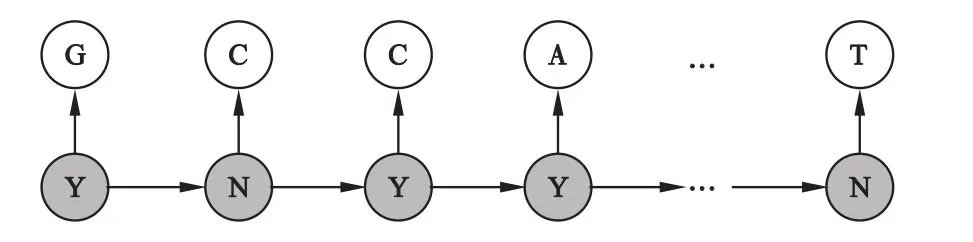
图1 序列识别的HMM内部示意图
2.隐马尔可夫模型的三个基本问题
评估问题:根据给定模型求某个观察值序列发生的概率P(0|λ),用来评估模型和给定观察输出序列的匹配程度,从而达到在一系列候选对象中选取最佳的匹配对象。常用算法为向前/向后算法,该算法可以有效地减少计算量[6-7]。
解码问题:在给定观察序列和模型的前提下,求产生观察值序列的最有可能的状态序列,常用算法为Viterbi算法。
学习问题:根据给定的观察值序列,调整模型参数,使其产生观察值序列的概率P(0|λ)最大。常用算法为Baum-Welch算法。
实例分析
数据来源于美国国家生物信息技术中心(NCBI),从共享资源中下载到已标识出编码区和非编码区的大肠杆菌全基因组序列。
1.隐马尔可夫模型的建立
采用Baum-Welch算法,利用Rstudio软件中的HMM包,对大肠杆菌编码区和非编码区序列分别进行训练建模:选取三分之二的大肠杆菌编码区序列共2750条建立HMM-gene模型,同样针对2330条大肠杆菌非编码序列建立HMM-nogene模型。迭代次数设置为100次,delta默认为1E-9,伪数默认为0。序列的状态集合为{编码区Y,非编码区N},观察集合为{A,T,C,G}。初始的转移概率矩阵A和发射概率B的定义如表1,训练后建立的模型结果如表2。

表1 隐马尔可夫模型初始参数

表2 隐马尔可夫模型参数
2.隐马尔可夫模型对DNA序列的识别结果
根据建立好的HMM-gene模型以及HMM-nogene模型,可以直接判断某位置上核苷酸的属性,但由于基因删除、插入等多种基因突变的出现,单一分析某一位点的属性显然是不够的,需要对一定长度序列的性质进行判断。分别统计在两模型下,被识别为编码状态的核苷酸与识别为非编码状态的核苷酸,即Y/N的比例,分别记为(1)和(2)。以两者的差作为特征指标,若差值大于0,则认为特征倾向于HMM-gene模型,判断序列为编码序列,若差值小于0,则识别为非编码序列,差值为0时,尚不能判断。该方法避免了复杂的计算,同时也避免了下溢现象,即由于HMM在计算概率时利用了条件概率思想,因此多次计算结果会出现无限趋于0的现象。
从余下的1/3序列中随机选取编码和非编码序列各180条作为测试数据集。表3,表4分别为利用HMM-nogene及HMM-gene模型对编码和非编码序列各180条的识别结果,当全部识别为Y时,为了避免分母为0,将N记为1。表5为编码和非编码序列识别结果比较,认为两者识别结果差别有统计学意义(χ2=10.840,P=0.028),其中,非编码序列中相持现象较多,可能与其结构有一定的相关性。
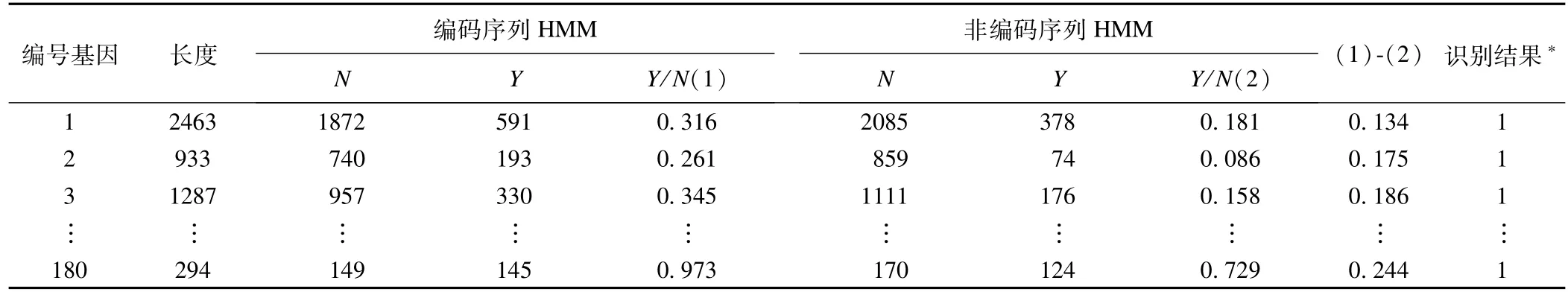
表3 编码序列识别结果

表4 非编码序列识别结果
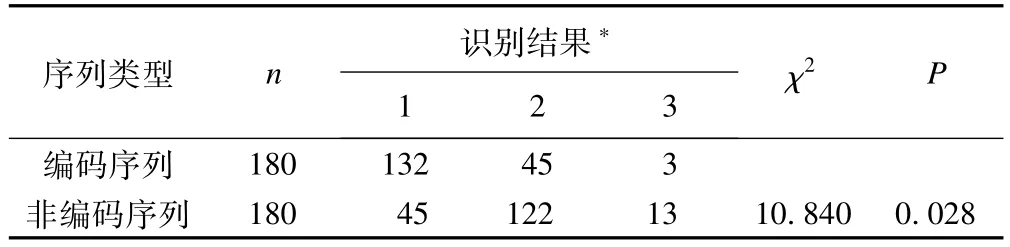
表5 编码序列和非编码序列识别结果比较
3.识别结果的评价
识别结果的评价采用敏感度Sn(sensitivity)、特异度Tn(specificity)及精确度Ac(accuracy)指标[8]。灵敏度,又称之为真阳性率,反映了正确识别出编码序列的能力。特异度,又称真阴性率,反映了正确识别出非编码序列的能力。精确度定义为特异度和灵敏度的均数。三者范围均在0~1之间,值越大,评价效果越理想。公式如下:

其中TP表示编码序列中被正确识别为编码序列的数目,FN表示编码序列中未被正确识别为编码序列数目,TN表示非编码序列中被正确识别为非编码序列,FP表示非编码训练中未被正确识别出来的非编码序列。
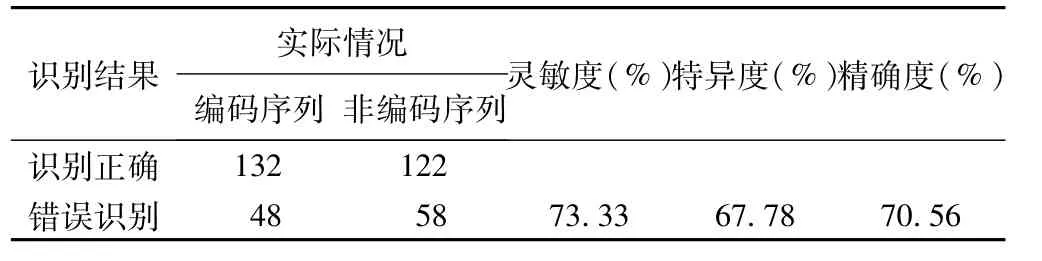
表6 对HMM模型识别序列的评价
讨 论
HMM不限制输入序列的长度,不需要指导者,有形成模块或层次结构进而形成完整的模型识别系统的优点,非常适用于有状态转移、线性序列的数据,同时,HMM运算速度比支持向量机快,对于大样本的生物信息数据分析有很大的优势[3],故在基因识别、序列比对等生物信息领域应用广泛。
但是,本文中HMM对原核生物编码序列的识别精确度不够理想,小于同类研究[3],究其原因如下:(1)对训练序列作预处理时,仅剔除了序列长度小于80bp或大于20000bp的序列,未采取其他措施控制训练序列的质量,进一步可选取等长度、较短序列进行训练,以研究训练序列长度对识别结果的影响;(2)未考虑原核生物的重叠信息,进一步应对两条基因的重叠序列进行处理,以增大识别精度;(3)一阶隐马尔可夫模型有三个条件独立基本假设,即状态独立性、观察独立性、状态与具体时间无关,事实上,在此刻出现的观测输出概率不仅依赖于系统前一状态,也很可能同之前的系统状态有关,因此进一步可在HMM的基础上进行碱基相关性的计算[9]。
总之,本文通过对原核生物序列进行训练后建立HMM,对DNA序列进行了识别,用特异度、灵敏度以及精确度三个指标进行了评价,为生物序列识别提供了方法学参考。今后可进一步研究HMM在序列比对中的应用,以及研究HMM对真核生物序列以及蛋白质序列的识别。
1.Goel N,Singh S,Aseri TC.A Review of Soft Computing Techniques for Gene Prediction.ISRN Genomics,2013,2013.
2.房颖.基于统计的基因识别算法研究[硕士论文].长春:吉林大学,2007.
3.罗泽举,李艳会,宋丽红,等.基于隐马尔可夫模型的DNA序列识别.华南理工大学报(自然科学版).2007,135(8):123-126.
4.Stormo G.Gene-finding approaches for eukaryotes.Genome research,2000,10(4):394-397.
5.潘海燕,丁元林,胡利人,等.隐Markov模型及其在慢性病流行病学研究中的应用.中国卫生统计,2009,26(1):38-40.
6.Baum LE,Egon JA.An inequality with applications to statistical estimation for probabilistic functions of a Markov process and to amodel for ecology.Bull Amer,Meterol,SOC,1967,73:360-363.
7.Baum LE,Sell GR.Grow th functions for transformations on manifolds. Pac,J,Math,1968,27(2):211-227.
8.Tompa M,Li N,Bailey TL,et al.Assessing computational tools for the discovery of transcription factor binding sites.Nat Biotechnol,2005,1(23):137-144.
9.张爽.基于HMM的转录因子结合位点识别方法研究[硕士学位论文].沈阳:东北师范大学,2009.
(责任编辑:刘壮)
The Coding Sequence Recognition of Prokaryote Based on Hidden Markov Model
Cao Hongyan,Ma Jing,Li Zhi,et al.(School of Public Health,Shanxi Medical University(030001),Taiyuan)
ObjectiveTo explore the identification of Escherichia coli coding sequence with Hidden Markov Model,so as to provide methods for the research of mining biological information and pathogenic loci.MethodsWe train the data set of Escherichia coli to model and identify the test set,and then evaluate the results using specificity,sensitivity and accuracy.ResultsThe specificity is 67.78%,the sensitivity is 73.33%and the accuracy is 70.56%based on the method of the paper.ConclusionHidden Markov Model can simulate the transformation of the discrete state very well,applied to identify the data of transformation state and linear sequence.
Hidden markov model;Coding region recognition;Escherichia coli
*:国家自然科学基金资助项目(31071156)
△通信作者:张岩波,Email:yanbozh@126.com
猜你喜欢
小学生学习指导(中年级)(2021年12期)2021-12-30
汉字汉语研究(2020年2期)2020-08-13
电子制作(2019年22期)2020-01-14
计算技术与自动化(2019年3期)2019-11-05
小学生作文(低年级适用)(2019年5期)2019-07-26
智富时代(2019年6期)2019-07-24
智富时代(2019年6期)2019-07-24
疯狂英语·新读写(2018年3期)2018-11-29
读友·少年文学(清雅版)(2018年12期)2018-04-04
山东青年(2016年3期)2016-02-28
