
三(3,5-二甲基苄基)氯化锡和四(间氰基苄基)锡的合成、晶体结构和量子化学研究
2015-04-01 06:16王剑秋张复兴邝代治冯泳兰庾江喜蒋伍玖朱小明
无机化学学报 2015年2期
王剑秋 张复兴邝代治 冯泳兰 庾江喜 蒋伍玖 朱小明
(衡阳师范学院化学与材料科学系,功能金属有机材料湖南省普通高等学校重点实验室,衡阳421008)
三(3,5-二甲基苄基)氯化锡和四(间氰基苄基)锡的合成、晶体结构和量子化学研究
王剑秋 张复兴*邝代治 冯泳兰 庾江喜 蒋伍玖 朱小明
(衡阳师范学院化学与材料科学系,功能金属有机材料湖南省普通高等学校重点实验室,衡阳421008)
以3,5-二甲基苄基氯和间氰基苄基氯在适当的溶剂中与锡粉反应,合成了三(3,5-二甲基苄基)氯化锡(1)和四(间氰基苄基)锡(2),经X射线衍射方法测定了化合物的晶体结构。化合物1属单斜晶系,空间群为P21/m,晶体学参数:a=0.584 03(4)nm,b=1.966 37(14)nm,c=0.856 46(5)nm,β=95.138(3),V=0.979 62(11)nm3,Z=2,Dc=1.735 g·cm-3,μ(Mo Kα)=14.53 cm-1,F(000)= 524,R1=0.043 7,wR2=0.123 2。化合物2属单斜晶系,空间群为C2/c,晶体学参数:a=1.692 21(12)nm,b=1.167 41(8)nm,c= 1.539 41(11)nm,β=116.615(10)°,V=2.718 9(3)nm3,Z=4,Dc=1.424 g·cm-3,μ(Mo Kα)=9.67cm-1,F(000)=1 176,R1=0.017 5,wR2= 0.046 1;中心锡原子为畸变四面体构型。对其结构进行量子化学从头计算,探讨了配合物的稳定性、分子轨道能量以及部分前沿分子轨道的组成特征。
三(3,5-二甲基苄基)氯化锡;四(间氰基苄基)锡;合成;晶体结构
烃基锡化合物由于具有结构的多样性、丰富的反应性、较强的生物活性和催化活性,多年来一直引起人们的兴趣[1-5]。烃基锡化合物有多种类型,其中最基础的是烃基卤化锡,其它都可视为其衍生物。烃基卤化锡又有一烃基三卤化锡、二烃基二卤化锡和三烃基卤化锡,最重要和最常见的是二烃基二卤化锡和三烃基卤化锡。除此之外,还有四烃基锡。由于取代苄基结构的多样性和独特的反应性,自上世纪九十年代初我们开创了取代苄基锡化合物的研究以来,人们对取代苄基锡的研究一直有较高的关注,合成了一系列取代苄基锡化合物[6-13]。在研究中人们发现2种有趣的现象:
一是在相同的反应条件下,苯环上连有的取代基的性质和取代基的位置对取代苄基锡形成和结构有明显的影响。如在一定的条件下,当取代基为卤素时,利用邻位取代苄基卤做原料和锡反应,主要得到三邻卤苄基卤化锡,而利用对卤苄基卤做原料则主要得到四对卤苄基锡[9-11],但当取代基为氰基时,对位取代的却得到三(对氰基苄基)氯化锡[12]。二是同一取代苄基卤在不同的条件下与锡反应,可得到不同的取代苄基锡,如对氰基苄基卤在不同的条件下与锡反应,可得到三(对氰基苄基)氯化锡[12]和四(对氰基苄基)锡[13]。为了更进一步探索取代苄基锡类化合物的反应条件和取代基的类型对产物的影响,我们在一定的条件下利用3,5-二甲基苄基氯和间氰基苄基氯与锡粉反应,合成了三(3,5-二甲基苄基)氯化锡(1)和四(间氰基苄基)锡(2),并通过元素分析、红外光谱对其结构进行了表征。用X-射线单晶衍射测定了该化合物的晶体结构,对其结构进行量子化学从头计算,探讨了配合物分子的稳定性、分子轨道能量以及一些前沿分子轨道的组成特征。
1 实验部分
1.1 试剂和仪器
IR用日本岛津FTIR-8700红外光谱仪(4 000~400 cm-1,KBr压片)测定,元素分析用PE-2400(Ⅱ)元素分析仪测定,晶体结构用Bruker SMART APEXⅡCCD衍射仪测定,熔点用北京产XT4双目体视显微熔点测定仪测定,温度计未经校正。所有试剂均为化学纯。
1.2 化合物的合成
化合物1的合成:在150 mL的三颈烧瓶中加入15.55 g(0.1 mol)3,5-二甲基苄基氯,5.93 g(0.05 mol)锡粉和100 mL正丁醇,少量碘,搅拌下将1 g镁条分批加入。快速搅拌回流6 h,趁热滤出未反应的锡粉,滤液倒入适量的稀盐酸溶液中,充分搅拌,冷却后分出水层。将正丁醇减压浓缩至适当的体积后放置,析出白色固体。分出固体,用苯重结晶得无色三(3,5-二甲基苄基)氯化锡11.95 g,收率70.13%。m.p.94~95℃。元素分析(C27H33ClSn)(%):实验值(计算值):C 62.94(63.38),H 6.54(6.46)。红外光谱主要吸收峰(ν/cm-1):3 053(w),2 974(w),1 611(s),1 594 (s),1 479(m),1 443(m),557(w),519(w),438(m)。
化合物2的合成用15.2 g间氰基苄基氯代替3,5-二甲基苄基氯,按照合成化合物1的方法,得四(间氰基苄基)锡无色晶体10.08 g,收率69.18%,m.p. 120~121℃。元素分析(C32H24N4Sn):实验值(计算值,%):C 65.98(65.90),H 4.16(4.19),N9.56(9.61)。红外光谱主要吸收峰(ν/cm-1):3 055(w),2 972(w),2228(m),1 591(s),1 576(m),1 477(m),1 425(w),565 (w),502(m),440(m)。
1.3 晶体结构分析
分别选取大小为0.23 mm×0.21 mm×0.19 mm和0.25 mm×0.21 mm×0.19 mm的晶体,在Bruker SMART APEXⅡCCD单晶衍射仪上,采用经石墨单色化的Mo Kα射线(λ=0.071 073 nm),于296(2) K,以φ~ω扫描方式收集数据。可观察衍射点分别为2 089个和2 577个[I>2σ(I)]用于结构分析和精修。全部数据经Lp因子和经验吸收校正。晶体结构由直接法解出,非氢原子坐标通过数轮差值Fourier合成陆续确定,理论加氢法给出氢原子在晶胞中的位置坐标。对非氢原子坐标及其各向异性热参数进行全矩阵最小二乘法修正。全部结构分析计算工作采用SHELX-97程序[14]系统完成。晶体学数据详见表1。
CCDC:1030252,1;1030251,2。
2 结果与讨论
2.1 化合物的晶体结构
化合物的主要键长和键角分别列于表2、3,化合物的分子结构见图1、图2。
化合物1:由分子结构图和结构参数可知,化合物为单体结构,中心锡原子与3个亚甲基碳和1个氯原子相连,形成四面体构型。分子是以经过Cl、Sn、C(10)、C(11)、C(14)5个原子的平面为对称面的面对称分子,扭转角∠Cl(1)-Sn(1)-C(10)-C(11)为0°。Sn-C键Sn(1)-C(1)和Sn(1)-C(1i)的键长均为0.213 6 nm,Sn(1)-C(10)的键长为0.214 2(7)nm,亚甲基碳与中心锡原子间的键角∠C(1)-Sn(1)-C(1i)为118.10°,∠C(1i) -Sn(1)-C(10)和∠C(1)-Sn(1)-C(10)均为114.57°,都比正四面体键角大;Sn-Cl键键长为0.237 85 nm,CSn-Cl键的键角∠C(10)-Sn(1)-Cl(1)为102.9°,∠C(1)-Sn(1)-Cl(1)和∠C(1i)-Sn(1)-Cl(1)均为101.68°,都比正四面体角小。这种空间排列决定了中心锡原子与亚甲基碳原子和氯原子构成一个畸变的四面体构型。

表1 化合物的晶体学数据Table 1Crystallographic data of compounds 1 and 2

表2 化合物的部分键长Table 2Selected bond lengths(nm)for compounds 1 and 2

表3 化合物的部分键角(°)Table 3Selected bond angles(°)for compounds 1 and 2

图1 化合物1的分子结构图(椭球概率30%)Fig.1Molecular structure of compound 1 with the ellipsoids drawn at the 30%probability level

图2 化合物2的分子结构图(椭球概率30%)Fig.2Molecular structure of compound 2 with the ellipsoids drawn at the 30%probability level
化合物2:由分子结构图和结构参数可知,化合物的晶体结构中,锡原子与4个苄基的亚甲基连接,形成四面体构型,分子中存在二重对称轴。Sn-C键键长分别为0.216 40和0.216 44 nm。亚甲基碳与中心锡原子间的键角分别为∠C(1)-Sn(1)-C(1i)= 113.08(10)°、∠C(9i)-Sn(1)-C(1)=110.91(7)°、∠C(1)-Sn(1)-C(9)=106.12(7)°、∠C(1i)-Sn(1)-C(9i)=119.91(7)°、∠C((1i)-Sn(1)-C(9)=110.91(7)°、∠C(9)-Sn(1)-C(9i)= 109.73(10)°,均偏离了正四面体角。因此,中心锡原子为畸变的四面体构型。化合物2的Sn-C键键长比四(对氰基苄基)锡的Sn-C键平均键长(0.216 575 nm)[13]要短,这体现了取代基在苯环上的位置的影响。
2.2 量子化学研究
2.2.1 分子的总能量和前沿分子轨道能量
根据晶体结构的原子坐标,运用Gaussian 03W程序和B3ylp/lanl2dz基组水平,计算得到分子的总能量和前沿分子轨道能量。配合物1:ET= -1 066.129 728 9 a.u.,EHOMO=-0.204 27 a.u.,ELUMO= -0.019 59 a.u.,ΔELUMO-HOMO=0.184 68 a.u.;配合物2:ET=-1 455.459 141 3 a.u.,EHOMO=-0.257 77 a.u.,ELUMO=-0.065 19 a.u.,ΔELUMO-HOMO=0.192 58 a.u.。从体系能量和前沿轨道的能量分析,2个化合物总能量和占有轨道能量均较低,表明2个化合物分子结构稳定。最高占据轨道与最低未占轨道的能量间隙ΔE均较大,从氧化还原转移的角度分析,均较难失去电子而被氧化。
2.2.2 轨道成分分析
为探索化合物的电子结构与成键特征,对化合物分子轨道进行分析,用参与组合的各类原子轨道系数的平方和来表示该部分在分子轨道中的贡献,并经归一化。
配合物1:把化合物原子分为4部分:(a)锡原子Sn;(b)氯原子Cl;(c)碳原子C;(d)氢原子H。前沿占有轨道和未占有轨道各取5个,计算结果如表4和图3所示。
表4和图3显示配合物1分子的成键特征:①前沿占有分子轨道中,二甲基苄基碳原子对分子轨道的贡献最大,为90.82%,并且在深层次轨道中均有较大的贡献,说明:一是苯环具有良好的共轭离域性和稳定性;二是苯环上的甲基和亚甲基的超共轭效应使苄基稳定;三是亚甲基碳原子有较大的贡献,因此其与锡原子结合较牢固,Sn-C键较稳定。②前沿占有分子轨道中,氯原子对分子轨道贡献很小,为0.83%,并且在深层次轨道中的贡献也很小,说明Sn-Cl键的稳定性有一定的限度。③比较HOMO与LUMO的各类原子轨道成份,可以看出,当电子从HOMO激发到LUMO轨道时,主要是苄基上的电子向锡原子转移。

表4 化合物1的分子轨道组成(%)Table 4Some calculated frontier molecular orbitals composition of compound 1
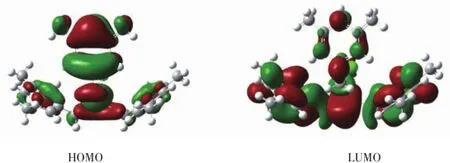
图3 化合物1的前沿分子轨道示意图Fig.3Schematic diagram of frontier MO for compound 1
配合物2:把化合物原子分为4部分:(a)锡原子Sn;(b)氮原子N;(c)碳原子C;(d)氢原子H。前沿占有轨道和未占有轨道各取5个,计算结果如表5和图4所示。
表5和图4显示化合物2分子的成键特征:①前沿占有分子轨道中,碳原子对分子轨道的贡献最大,达90.77%,并且在深层次轨道中均有较大的贡献,其中主要来自苯环碳原子,表明苯环具有良好的共轭离域性和稳定性。②前沿占有分子轨道中,氰基氮原子对分子轨道贡献较小,为1.53%,并且在深层次轨道中的贡献较小,这可能是由于氰基的极性,使其稳定性有一定的限度,亦表明氰基与苯环共轭性有限。③比较HOMO与LUMO的各类原子轨道成份,可以看出,当电子从HOMO激发到LUMO轨道时,主要是锡原子上的电子通过苄基向氰基转移,电子转移的结果表明分子在激发态时Sn-C键的强度减弱,化合物的稳定性降低。

表5 化合物2的分子轨道组成(%)Table 5Calculated some frontier molecular orbitals composition of complex 2

图4 化合物2的前沿分子轨道示意图Fig.4Schematic diagram of frontier MO for compound 2
[1]Chandrasekhar V,Thirumoorthi R,Metre R K,et al.J. Organomet.Chem.,2011,696:600-606
[2]Effendy,Marchetti F,Marinelli A,et al.J.Inorg.Chem. Acta,2011,366:388-393
[3]HanifM,HussainM,AliS,etal.Polyhedron,2010,29:613-619
[4]ZHANG Xiao-Yan(张晓燕),YANG Guang(杨光),ZHANG Jun(张俊),et al.Chem.J.Chinese Universities(高等学校化学学报),2010,31(6):1162-1166
[5]Siddiqi Z A,Shahid M,Kumar S,et al.J.Organomet. Chem.,2009,694:3768-3774
[6]YIN Han-Dong(尹汉东),GAO Zhong-Jun(高中军),XU Hao-Long(徐浩龙),et al.Chinese J.Inorg.Chem.(无机化学学报),2006,22(2):371~374
[7]YIN Han-Dong(尹汉东),HONG Mei(洪敏),WANG Chuan-Hua(王传华).Chinese J.Inorg.Chem.(无机化学学报), 2005,21(5):713~715
[8]ZHANG Fu-Xing(张复兴),WANG Jian-Qiu(王剑秋), KUANG Dai-Zhi(邝代治),et al.Chinese J.Inorg.Chem. (无机化学学报),2004,20(2):202~204
[9]WANG Jian-Qiu(王剑秋),ZHANG Fu-Xing(张复兴), KUANG Dai-Zhi(邝代治),et al.Chinese J.Org.Chem.(有机化学),2004,24(1):80~84
[10]ZHANG Fu-Xing(张复兴),WANG Jian-Qiu(王剑秋), KUANG Dai-Zhi(邝代治),et al.Chinese J.Org.Chem.(有机化学),2003,23(4):368~371
[11]ZHANG Fu-Xing(张复兴),KUANG Dai-Zhi(邝代治),XU Zhi-Feng(许志锋),et al.Chinese J.Inorg.Chem.(无机化学学报),2002,18(8):854~858
[12]WANG Jian-Qiu(王剑秋),KUANG Dai-Zhi(邝代治), ZHANG Fu-Xing(张复兴),et al.Chinese J.Inorg.Chem. (无机化学学报),2003,19(10):1109~1112
[13]YIN Han-Dong(尹汉东),GAO Zhong-Jun(高中军),XU Hao-Long(徐浩龙),et al.Chinese J.Inorg.Chem.(无机化学学报),2005,11(21):1743-1746
[14]Sheldrick G M.SHELXTL,Version 5.03.Madison,Wisconsin, USA:Siemens Analytical X-ray Division,1994.
Syntheses,Crystal Structures and Quantum Chemistry of Tri(3,5-dimethylbenzyl)tin Chloride and Tetra(m-cyanobenzyl)tin
WANG Jian-QiuZHANG Fu-Xing*KUANG Dai-ZhiFENG Yong-Lan YU Jiang-XiJIANG Wu-JiuZHU Xiao-Ming
(Key Laboratory of Functional Organometallic Materials of College of Hunan Province,Department of Chemistry and Material Science, Hengyang Normal University,Hengyang,Hunan 421008,China)
The tri(3,5-dimethylbenzyl)tin chloride(1)and the tetra(m-cyanobenzyl)tin(2)have been synthesized. The crystalstructures of the complexes were determined by X-ray diffraction.The crystal of 1 belongs to monoclinic space group P21/m with a=0.584 03(4)nm,b=1.966 37(14)nm,c=0.856 46(5)nm,β=95.138(3)°, V=0.979 62(11)nm3,Z=2,Dc=1.735 g·cm-3,μ(Mo Kα)=14.53 cm-1,F(000)=524,R1=0.043 7,wR2=0.123 2.The crystal 2 belongs to monoclinic space group C2/c with a=1.692 21(12)nm,b=1.167 41(8)nm,c=1.539 41(11) nm,β=116.615(10)°,V=2.718 9(3)nm3,Z=4,Dc=1.424 g·cm-3,μ(Mo Kα)=9.67 cm-1,F(000)=1 176,R1=0.017 5, wR2=0.046 1.The tin atoms have a distorted tetrahedral geometry.The stabilities,some frontier molecular orbital energies and composition characteristics of some frontier molecular orbital of the complex have been investigated by quantum chemistry calculation.CCDC:1030252,1;1030251,2.
tri(3,5-dimethylbenzyl)tin chloride;tetra(m-cyanobenzyl)tin;synthesis;crystal structure
O614.43+2
A
1001-4861(2015)02-0237-06
10.11862/CJIC.2015.018
2014-06-19。收修改稿日期:2014-10-20。
湖南省高校创新平台开放基金项目(No.12K124)、湖南省自然科学基金项目(No.11JJ3021)、湖南省重点学科基金和湖南省高校重点实验室开放基金(No.13K02)资助。
*通讯联系人。E-mail:zfx8056@163.com;Tel:0734-8484932
猜你喜欢
河南化工(2021年4期)2021-05-12
昆明医科大学学报(2021年2期)2021-03-29
上海计量测试(2020年1期)2020-03-18
四川警察学院学报(2019年6期)2019-12-28
文化产业(2016年6期)2016-10-19
浙江大学学报(工学版)(2016年2期)2016-06-05
药学研究(2015年11期)2015-12-19
中国洗涤用品工业(2015年9期)2015-02-28
中国塑料(2014年10期)2014-10-17
天然产物研究与开发(2014年6期)2014-04-27
