
Rethinking bioequivalence and equivalence requirements of orally inhaled drug products
2015-05-16 07:55DinAlNumniPhilippeColucciMurryDuchrme
Din Al-Numni,Philippe Colucci,Murry P.Duchrme,b,*
aLearn and Con fi rm Inc.,St-Laurent,QC,Canada
bFaculté de Pharmacie,University of Montreal,Montreal,QC,Canada
Review
Rethinking bioequivalence and equivalence requirements of orally inhaled drug products
Dina Al-Numania,Philippe Coluccia,Murray P.Ducharmea,b,*
aLearn and Con fi rm Inc.,St-Laurent,QC,Canada
bFaculté de Pharmacie,University of Montreal,Montreal,QC,Canada
ARTICLE INFO
Article history:
Received 16 July 2015
Accepted 2 August 2015
Available online 24 August 2015
Orally inhaled drug products(OIPs),such as corticosteroids and bronchodilators,are at the forefront of asthma and chronic obstructive pulmonary disease treatments,two diseases that af fl ict worldwide populations.Introducing generics of these products is essential,as the pricing of these medications remain a barrier to adequate patient care.Currently,there is no consensus between regulatory bodies as to the bioequivalence and equivalence requirements of OIPs that are intended for local action in the lungs.This manuscript critically reviews these requirements and presents future directions for clinicians,scientists,and regulators to consider to optimize the development and approval of OIPs.
Inhalation products
Pharmacokinetics
Pharmacodynamics
Bioequivalence
Therapeutic equivalence Clinical endpoint studies
1. Introduction
Drug delivery directly to the lung of patients with air fl ow obstructions allows the delivery of relatively small doses of drug directly to the airway achieving high local concentrations,while minimizing systemic adverse drug effects[1].Inhalers are most commonly prescribed in the treatment of asthma and chronic obstructive pulmonary disease(COPD)[2].
Asthma has a prevalence ranging from 1 to 18%and affects approximately 300 million individuals worldwide[3].Inhaled corticosteroids(ICS),such as fl uticasone,are one of the cornerstones in the treatment of moderate to severe asthma[4]. They play a critical role in the long-term control of asthma through their anti-in fl ammatory effects.They have been shown to reduce asthma symptoms[5],improve quality of life[5], improve lung function[5],decrease airway hyper responsiveness[6],control airway in fl ammation[7],reduce the frequency and severity of exacerbations and reduce asthma-related mortality[8].
Inhaled bronchodilators are also used in the treatment of asthma[4]and are central in the treatment of patients with COPD[9].COPD has a prevalence ranging from 7.8 to 19.7% world-wide,with a range of 3 to 11%among never smokers [10].The relaxation of airway smooth muscles that lead to bronchodilation can be achieved by inhibiting acetylcholine signaling via muscarinic M3receptors expressed on airway smooth muscle.This can be performed with a muscarinic antagonist (e.g.,tiotropium)or by stimulating beta-2 adrenoceptors with a beta-2 agonist(e.g.,salmeterol)[9].
In light of the critical role of ICS and inhaled bronchodilators in the treatment of asthma and COPD,it is not surprising that there is considerable interest in developing new combinations of existing products,follow-ons and/or generic products of existing ICS and inhaled bronchodilators.The use of generic drugs in general is on the rise,accounting for 51%of prescriptions in 2002 and 67%in 2007 in the US[11].Recent estimates indicate generic drugs account for approximately 80%of all retail prescriptions dispensed in the US[12],and 66%in Canada [13].These growing trends,coupled with the urgent need to relieve the economic burden of prescription drugs on patients and healthcare providers,force us to reconsider how the drug development of generic ICS and inhaled bronchodilators can be improved to bring such products to market in a safe and timely manner.This puts additional pressure on innovative companies to bring new products on the market as their existing ones become preys to generic competition.With new products costing an average of one to two billion dollars[14], companies are not only developing new products entirely but have also repositioned old products by introducing new and improved devices(e.g.,Diskus®line of products)or combination products(e.g.,Advair®).
The aim of this manuscript is to review current regulatory recommendations for highly regulated regions(e.g.,Health Canada(HC),US Food and Drug Administration(FDA),European Medicines Agency(EMA))for the development of followon and/or generic inhaler products,critically examine assumptions and principles that underlie some of these recommendations,focusing on pharmacological principles,and suggest other approaches that may be considered in the future.
2. General bioequivalence requirements for OIPs:local vs systemic action
Clinical regulatory requirements to ful fi ll in order to obtain an approval for a generic or follow-on drug product typically depend fi rst on the relationship between the systemic circulation and the assumed site of activity of the active pharmaceutical ingredient(API).For drug products reaching fi rst the systemic circulation and then distributing to the theoretical site(s)of activity via the systemic circulation, pharmacokinetic(PK)studies indicating equivalence in terms of systemic exposure(e.g.,typically Cmaxand AUC)between a follow-on/generic formulation and a reference one will ensure that their safety and ef fi cacy will be similar because a similar systemic exposure will result in similar exposures at the theoretical site(s)of activity.For these products,a generic version would be demonstrated to be bioequivalent to a reference product using one or more PK equivalence studies.A theoretical example of such a product would be a potential generic inhaled insulin product,as insulin will reach its sites of activity via the systemic circulation.
On the other hand,a drug product for which its API is delivered from a formulation at its intended site(s)of activity before reaching the systemic circulation is typically called a“locally acting”product.For these products equivalence in terms of systemic exposure may not ensure“local”exposure equivalence and therefore a PK equivalence study may not suf fi ce to ensure similar ef fi cacy and safety.Examples of“locally”acting products within the lungs include inhalers for COPD and asthma,such as albuterol,budesonide,ipratropium,tiotropium, fl uticasone and salmeterol.
3. Current regulatory requirements for generic submissions of“locally acting”inhaled products
3.1. North American requirements (FDA and HC)Although clear guidelines exist in the U.S.with respect to the conduct of bioequivalence(BE)studies of OIPs[15],presently there is no general guideline on how to establish BE of OIPs.
In the US,the FDA has issued a general guidance documenting the CMC requirements[16],and three individual product BE guidances:one for budesonide suspension inhaler [17],one for albuterol sulfate metered dose inhaler[18],and one for the fl uticasone propionate/salmeterol xinafoate dry powder inhaler[19].
Despite the absence of an FDA guidance document on the BE of OIP in general,scienti fi c and regulatory considerations for demonstrating BE between dry powder inhalers(DPI)have been proposed[20],and recently reviewed[21].The FDA has developed an aggregate weight-of-evidence approach which utilizes in vitro studies,PK equivalence studies,and pharmacodynamic(PD)or clinical endpoint(CE)studies(also called Therapeutic Equivalence(TE)studies),to establish BE of inhalation products[21].Equivalence in all categories of interest is required[22].As such,a generic formulation of a reference inhaler product needs to demonstrate that 1)its device will be judged to be equivalent in a patient’s hand,2)that its in vitro characteristics and performance in terms of emitted dose and aerodynamic particle size distribution is equivalent,3)that its systemic PK exposure is equivalent,and fi nally that 4)its clinical ef fi cacy is equivalent.
At the present moment,the requirements for HC are very similar to those of the US FDA.HC also demands that equivalence be demonstrated in terms of device,in vitro characteristics and performance,systemic PK exposure,and clinical ef fi cacy. In Canada,the requirements are speci fi ed in three guidances,one specifying the requirements for second entry shortacting beta-2 agonist metered dose inhalers[23],one focusing on the in vitro requirements for nasal and inhalation products[24],and fi nally a draft guidance specifying what studies would be needed to prove similar clinical safety and ef fi cacy [25].This last guidance was however recently pulled from the HC website,possibly an indication that a new updated version is forthcoming.One difference with the FDA is that HC is less prescriptive in terms of study design for the clinical equivalence study and sponsors can theoretically use different designs as long as they can prove to the agency that it will ensure equivalent ef fi cacy between the generic and reference inhaled products[25].
3.2. European requirements(EMA)
Many guidelines exist to describe what the requirements are for EU submissions.The guideline CPMP/EWP/4151/00 Rev.1covers the overall requirements and speci fi es the needed clinical studies[26],while the guideline EMEA/CHP/QWP/49313/ 2005 Corr.speci fi es what the in vitro requirements are[27].
The EMA guidelines allow the theoretical approval on the basis of in vitro data only provided that results are all favorable.If an applicant cannot demonstrate complete in vitro similarity,or if the applicant’s product is deemed too different fromthereferenceproduct,thenPKstudiestoestablishequivalent safety and ef fi cacy are suggested.Equivalence in terms of ef fi cacy is typically recommended to be established via a PK systemic exposure equivalence study where charcoal is administered to block GI absorption so that only the exposure of theAPIabsorbedviathelungiscompared.Equivalenceinterms of safety is done via a PK systemic exposure equivalence study but where charcoal is not administered,so that the total systemic exposure of the generic versus the reference product are compared,not just what is absorbed via the lung.Should PK studies fail or should they be considered to not address the speci fi c in vitro failures of the test product,clinical studies are then needed to establish equivalent safety and ef fi cacy.One important difference versus FDA or HC is that in Europe,OIPs are not approved as generic submissions but as hybrids,and they are not automatically substituted.Substitution is determined nationally.Very few international approvals appeared to have been granted since the issuing of the general guidance.The publicly available assessment reports suggest that not all regulators adhere strictly to all parts of the guideline.
It can be appreciated that the device and in vitro requirements are generally aligned between these three regulatory jurisdictions,but they differ substantially in how similar clinical safety and ef fi cacy must be demonstrated.The general requirements are summarized and contrasted in Table 1.
We can see that the requirements for HC and US submissions are relatively similar,while they are very different in the EU.For this latter one,de fi nite proof of device and in vitro similarity can theoretically suf fi ce for a“hybrid”submission, although it appears in practice that EU regulators are feeling more comfortable if sponsors do a minimum of two Phase I PK studies,with and without charcoal concomitant administration.A“practical”requirements table can be constructed(see Table 2)using the example of the Advair Diskus®product.
4. Current controversial aspects related to locally acting inhaled products
4.1. The need for concomitant charcoal administration in PK equivalence studies
One study design aspect that not all regulatory agencies agree with is the requirement by the EMA to perform two PK equivalence studies if all in vitro tests are not conclusive of equivalence; one study with and one study without charcoal.The PK study without charcoal is performed to prove“safety equivalence”as the total systemic exposure from both the lung and gut absorptions will be measured.The charcoal PK study is performed to prove“ef fi cacy equivalence”and will only measure the“lung absorption exposure”as the absorption from the gut will be inhibited by the charcoal.The theory for the charcoal study is that the systemic exposure from this study serves as a surrogate for the local lung absorption and exposure.This however ignores the fact that ef fi cacy can also be due to systemic free drug exposure resulting from the part of the inhaled dose that is swallowed and absorbed through the gut.Of course this will be virtually nil for low oral gut bioavailability and highly protein bound drugs like fl uticasone( fl uticasone has 1%oral gut bioavailability and is 99%plasma protein bound[28]),but it may be somewhat important for others such as albuterol orlevalbuterol,drugs that are very little plasma protein bound (8%)and that have high oral gut bioavailability(50%)[29,30].

Table 1-General regulatory agency requirements for demonstrating bioequivalence of generic OIPs.

Table 2-Potential minimum study requirements for generic submission of Advair Diskus®DPI.
In addition,it can be argued that the difference in systemic exposure due to the lung versus gut can easily be obtained using population PK modeling from the full exposure study performed without charcoal,and therefore the charcoal study could be avoided altogether because it is relatively easy to discriminate,using a population approach,the drug absorbed via the lung versus the gut[29].For products with a low gut bioavailability such as fl uticasone(<1%)[28],the systemic exposure anyways represents only the lung exposure as exposure due to gut absorption is negligible.For these products the charcoal study would provide essentially the same results as the study given without charcoal.For drugs where exposure due to gut absorption is expected to be signi fi cant (e.g.,exposure due to gut absorption accounting for let’s say more than 20%of total exposure)such as albuterol[31],then the plasma concentration-time pro fi le will typically show very clearly the dual absorption peaks with the fi rst peak appearing shortly after administration re fl ecting lung absorption and a second peak appearing later re fl ecting gut absorption.Population PK modeling can be performed to characterize the exposure associated with the lung and gut absorption separately,and determine if these different exposures are equivalent between products.The use of this population approach was previously reported by Auclair et al.[29].While it is reasonable for EMA to ask that PK equivalence be shown for lung absorption only(with charcoal)as well as lung and gut absorption(without charcoal),this could all be calculated from a single study without having to administer charcoal.
4.2. The need for dose-response for TE studies to be robustly discriminative
TE studies with CE are currently required by the US FDA and HC for a generic submission of OIPs.Based on the step-wiseapproach,the EU would require such studies only when the in vitro and/or PK studies do not show equivalence.A vital element of aTE study with CE is the demonstration of a doseresponse relationship,in order to con fi rm that a lack of difference in the CE is truly because the test and reference generate a similar level of effect,and not because the study lacks sensitivity in detecting a difference.This has been a major challenge,especially for products such as ICS,and is discussed further below.
An ideal biomarker for aTE study must possess several characteristics.It must be clinically relevant,i.e.,there must be a link between the biomarker and the pharmacological mechanism of the drug.It must also be objective,reproducible with a reversible effect that is dependent on the dose[32,33].As such, this would make it possible to establish a clear dose-response relationship and apply the dose-scale approach that is favored by the FDA[34].A biomarker that is ideal for one drug may not be ideal for another,as its relevance depends upon the drug’s mechanism of action,and in some cases,there may be no ideal biomarker at all.For example,forced expiratory volume in 1 second(FEV1)is a biomarker used to assess the response to beta-2 agonists[18],and it is an ideal biomarker as it is reversible and related to systemic drug concentrations,thus it exhibits a clear dose-response relationship.This biomarker has been used now for approximately 20 years for submissions of inhaled short-acting beta-2 agonists.FDA recently published an individual BE guidance for the beta-2 agonist albuterol that con fi rms its usage[18].
FEV1may not be a good choice of a biomarker for ICS, however,as it does not necessarily capture the dual mechanism of action of these drugs.The response to ICS occurs in two waves:the fi rst response occurring on Day 1 after the start of therapy and a latent response occurring 7 to 10 days later. One of the major mechanisms behind the anti-in fl ammatory effect of ICS in the treatment of asthma is via inhibition of the effects of pro-in fl ammatory cytokines following binding with glucocorticoid receptors[35].By contrast,endocrine and metabolic effects of corticosteroids are mediated through DNA binding[35].An ideal biomarker would be able to capture the spectrum of responses to ICS and maybe not just the early or late bronchodilatory response that is seen with FEV1.Regardless of this, fl uticasone and the combination product f l uticasone/salmeterol have been approved by many diverse regulatory agencies including the US FDA based on FEV1measures.Because of this,the FDA has published an individual BE guidance for the combination product fl uticasone/salmeterol and is asking generic sponsors to prove TE using FEV1measures after single dose administration and after 4 weeks of continued dosing[19].
Despite FDA issuing a BE guidance for the fl uticasone/ salmeterol inhaler combination product,there is still a need for a better biomarker for ICS response than FEV1[36,37].One potential biomarker that used to be suggested for many years by the FDA was exhaled nitric oxide(ENO).Although this biomarker was hoped to re fl ect the secondary(latent)mechanism of action of ICS,its response is in fl uenced by many factors, such as collection techniques,disease states,and other patient factors(food,drink,smoking)[38].Even though there is still some interest in using this biomarker for ICS[39],it is not necessarily recommended anymore by regulatory agencies as studies have revealed the absence of a dose-response relationship between ENO and fl uticasone administered at the marketed dosing regimens.The lowest labeled dose(88 mcg twice daily(BID))was associated with responses on or near the plateau of the dose-response curve and responses did not return to baseline levels after the 14-day washout period[40].Others have shown that ENO on its own cannot provide useful therapeutic information regarding response to ICS[41]or that it could be useful as an adjunct to guide therapy[42].
HC was recommending the use of sputum eosinophils for many years[25],believing that due to the presence of primed eosinophils in airways that contribute to in fl ammatory processes in asthmatics[37],this biomarker could characterize the anti-in fl ammatory activity of ICS.No consensus appears to exist though on the correlation between sputum eosinophils and ICS response.Some researchers have found a positive relationship between the two[43-49],while others have shown little or poor correlation[50,51].It is not clear if HC is still as optimistic regarding this biomarker for ICS as in the past,as the guidance document that contained it has been withdrawn from HC’s website.
Although biomarkers such as FEV1are generally accepted to evaluate response to bronchodilators(short-acting and longacting beta-2 adrenoreceptor agonists)[18,26],it can be argued that their utility for ICS response should be limited,because similarly to ENO,the smallest marketed dosing regimen of f l uticasone,for example,results in a response that is already at the top of the dose-response curve for FEV1.
Unfortunately no biomarker has been found or proposed to date that would enable a dose-response to be seen across the marketed doses of fl uticasone inhalers or other ICS.Ideally andassumingthatanadequatebiomarkercouldbefound,therapeutic equivalence would have to be demonstrated by proving that a dose-response relationship exists,otherwise the study may not be discriminative at all if the doses studied are already atthetopoftheresponsecurve.Thestudyshouldideallyinclude a placebo and assess a low dose of the drug(the lowest dose asrecommendedintheproductlabeling),aswellasahighdose (generally 2 to 4 times higher than the low dose).For the study to be valid,theoretically the response to the low dose of the drug must be superior to the placebo,and the response to the high dose of the drug must be greater than the response of thelowdose.Itiscriticaltobeabletoclearlydistinguishbetween responses to varying drug doses,otherwise it may be impossible to distinguish reliably between drug formulations.
With the accumulated experience we now have concerning the relationship between dose and response of PD parameters such as FEV1and ENO,simulations were made to understand the importance of the existence or not of a doseresponse on the resulting power and alpha error ofTE studies with CE[52].Using NONMEM®,thousands of TE studies each with a 5-way crossover design(placebo,low dose test,low dose reference,high dose test,high dose reference)were simulated.Different scenarios(i.e.,equivalent or non-equivalent product,varying test to reference ratios)were simulated.Each scenario included 1000 simulated TE studies,and each study had a sample size of 136 subjects completing the 5 way crossover.Equivalence was assessed using the dose-scale approach (Fig.1),in which the equivalence is calculated on the dose scale, and not on the response scale which is non-linear.
Simulations were performed when a dose-response truly exists(e.g.,the dose at which 50%(ED50)of the maximum effect (Emax)is equivalent to the lowest dose marketed)and when a dose-response does not exist,as in the case for ICS with FEV1, as the ED50is much lower than the lowest marketed dose. Results of the scenario where the dose-response is present are highlighted in Table 3.It can be seen that in the case where ED50occurs at approximately the low dose,such a TE design has adequate power(>80%)to conclude equivalence with the dosescaleapproachwhenindeedtheproductsare bioequivalent.When products are not bioequivalent,the alpha error is slightly increased,1 to 13.5%for a true test to reference ratio of 60 to 70%,but overall the products are shown to not be equivalent.
Results of the scenario where the dose-response is not present are highlighted in Table 4.This would be the case for example for fl uticasone using FEV1or ENO as the CE using the dose-scale,as the maximum effect is approximately seen with the lowest dose marketed.Results show that any TE study in this scenario using the dose-scale approach would have very little power(<20%)to conclude equivalence even when the products are truly equivalent.Results show that it would be almost impossible to prove equivalence and the method would not really distinguish between a bioequivalent product and a nonbioequivalent one,as the alpha error rates for non-bioequivalent products are virtually the same as the statistical power for bioequivalent products.
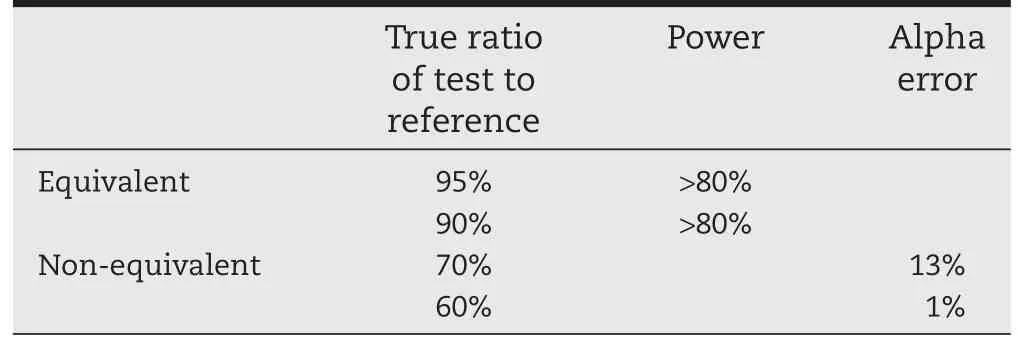
Table 3-Statistical power and alpha error of TE studies when a dose-response does exist and when the dose scale method is used to prove equivalence.

Table 4-Statistical Power and Alpha error of TE studies when a dose-response does not exist and when thedose scale method is used to prove equivalence.
These results show the importance of an existing doseresponse at the doses studied in a TE study with CE when the dose scale method is used.If the studied doses are at the top of the response curve,then the dose scale method unfortunately does not work and will almost never show equivalence between a test and a reference formulation.
4.3. When using the dose-scale approach should one study the high dose effect only for the reference or also for the generic product?
As presented earlier,the dose scale approach is a good method to assess TE in a study with CE when a dose-response truly exists at the dosing regimens studied.We have seen in Table 3 that with this method,and in an appropriately powered study, the power to prove equivalence is as expected when the test and reference products are equivalent,while when not equivalent the discriminatory power is also adequate although resulting in a slightly higher alpha error than what we typically see with PK equivalence studies.Therefore this method should be used whenTE studies with CE are performed versus the alternative of creating ratios and con fi dence intervals on the response scale which is not robust due to the non-linearity of the response.
FDA’s recommendations for a few products requiring the dose-scale approach are currently not always consistent.For albuterol inhalers,FDA is recommending a 4-way instead of a 5-way study[18].These are obviously minimum recommendations as a sponsor could always study more,but this study design includes a placebo,low dose test,low dose reference, and high dose reference.What is missing is the high dose of the test.Although not absolutely needed,as the dose-response curve can be assessed with reference doses only and equivalence is assessed on the low dose,we aimed at discovering the impact of including or not this fi fth arm in these studies.TenthousandTE studies with either 4-or 5-arm crossover designs including 136 patients each and for fi ve different scenarios (three equivalent products and two non-equivalent products) were simulated using NONMEM®.Results are presented in Table 5 and indicate that the statistical power to prove equivalence is as expected with the 5-arm design(approximately 80%), while it becomes much lower(approximately 60%)when a 4-arm design is selected and the test is not administered as a high dose.In order for the 4-arm design to be associated with 80%power,the study would have to include almost double thenumber of patients.Doubling the number of patients in a 4-arm study is much more complicated and costly than adding an additional arm.Pharmaceutical companies should therefore perform these studies as 5-arm designs instead of what is minimally recommended.

Table 5-Power of TE studies using the dose-scale approach with 4-arm(placebo,low dose test,low and high dose reference)versus 5-arm(adding high dose of the test)designs. Power Alpha error Equivalent 100% 79.8% 60.7% 95% 73% 57.4% 90% 67.7% 54.5% Non-equivalent 70% 24.3% 23.6% 60% 6.4% 10.6% True ratio of test to reference 5-arm design 4-arm design Power Alpha error
4.4. The need for TE studies once PK equivalence is demonstrated
Once a test and reference products are shown to be equivalent based on comparative PK studies,we are presenting an argument herein that TE studies using CE are no longer necessary.The basis of this argument is that 1)TE studies are not as discriminative as PK studies;2)the lungs should not be considered a“topical”organ per se,as it is one of the most highly perfused organ in the body because of its critical importance in oxygenating the body;and 3)a drug product can only act“locally”once it is in solution,not before,and once the API will appear in solution at the site of activity in the lung then its free drug concentrations should be immediately in equilibrium with the systemic circulation.
Daley-Yates and Parkins[53]summarized the in vitro,PK and PD studies of several inhaled products,and concluded that there was a lack of understanding between in vitro,PK and PD studies. The authors mentioned an example of a product that was found to be equivalent in terms of PK in one study[54],but not equivalent in terms of ef fi cacy(using FEV1)in another study(Kerwin et al.[55]).This comparison between two studies is completely misleading though,because the ef fi cacy study they referred to(Kerwin et al.[55])saw aligned differences between PK and ef fi cacy(i.e.,when PK was different the ef fi cacy was different).All other examples presented in the review by Daley-Yates and Parkins[53]showed that when PK was equivalent, ef fi cacy was always equivalent,but when PK was not equivalent,ef fi cacy was sometimes equivalent and sometimes not equivalent.This suggests that comparisons made using PD data when equivalence is assessed on the response scale lacks specif i city and with the tools we are using to date(e.g.,no doseresponse,proving equivalence with only one dose and with a biomarker that may not re fl ect the wide ef fi cacy of the OIP)it may not allow for a meaningful product discrimination for the assessment of equivalence and therefore one could argue that the study does not serve any true scienti fi c purpose.PK studies, on the other hand are much more sensitive and speci fi c to detect differences between two products.The ultimate question is,are OIPs acting“locally”within the lungs with their systemic exposure unrelated to their theoretical exposure therein?Lawrence et al.’s paper from 1998[56]is often quoted in this regard as offering evidence that much higher systemic exposure of fl uticasone following oral versus inhalation dosing does not result in ef fi cacy and therefore suggests that f l uticasone acts locally within the lungs with no equilibrium with the systemic circulation.We will see that this conclusion is incorrect in the next paragraphs.
The lung epithelium differs in cell type and thickness,with diminishing thickness from the trachea to alveoli.Following absorption via the inhalation route,an API reaching the lung epithelium must fi rst dissolve into a solution before it can act at the site of activity.Therefore,the exposure of the API at the site of activity in the lungs depends on the drug formulation characteristics,as well as physiologic ones[57].The lungs are the most or one of the most perfused organs of the body and these should not be considered as a topical site of absorption like the skin[36]but rather an extension of the systemic exposure because of this high perfusion.This permits drug in a solution state at the site of activity in the lung to be almost instantaneously distributed to the systemic circulation.This explains why maximum concentrations are seen virtually immediately in a few seconds and minutes after inhalation for drugs that have high solubility within the lungs.For example, in a previous study we have found that the absorption halflife of levalbuterol via inhalation into the systemic circulation in dogs was less than 2 minutes,virtually as fast as administering the drug via the intravenous route(29).The observed“total”systemic concentrations after lung absorption therefore should directly re fl ect the“active”unbound drug concentrations within the lungs.Drugs absorbed by the gut(e.g., tablet formulation)are accessible to the lung but only“unbound”drug will be available to be in equilibrium with the site of activity in the lung.As mentioned earlier,many argue that only local exposure from the lung is involved in the ef fi cacy of f l uticasone when concentrations following inhalation versus oral administrations are compared[56].This is an incorrect comparison because only“unbound”drug will equilibrate between the systemic circulation and the site of activity in the lungs, and therefore any comparison using total systemic concentrations(sum of fl uticasone bound and unbound to plasma proteins)after gut absorption will overestimate the exposure reaching the lung and consequently overestimate the expected ef fi cacy.This incorrect comparison can be shown with f l uticasone as presented by Lawrence et al.[56].
Fluticasone has an oral bioavailability that is less than 1% and its plasma protein binding is reported to be 99%[28].In the study reported by Lawrence et al.[56],no ef fi cacy wasobserved with a 20 mg oral administration of fl uticasone when compared to 100 and 500 mcg inhalation administrations.This was true even though systemic concentrations after oral administration were 2 to 3 times higher than after the 500 mcg inhalation administration.If we consider that total plasma concentrations after inhalation re fl ect the lung concentrations and that only unbound plasma concentration(less than 1%as protein binding is 99%)after oral administration re fl ects lung concentrations,then the lung fl uticasone exposure from the oral administration was approximately 50 times lower than that for the 500 mcg inhalation administration.In order to have equivalent lung exposures with the 100 and 500 mcg inhalation dose,the oral dose would have needed to be as high as 200 to 1000 mg instead of 20 mg.The importance of considering free drug concentrations and not total drug concentrations is further presented in Fig.2 for this particular fl uticasone example.
Fig.2 clearly indicates that because fl uticasone is 99%protein bound,a systemic overall exposure(AUC)of 629 pg.h/ml after inhalation dosing suggests that the AUC of fl uticasone in solution as free drug concentration at the site of activity in the lung was also 629 pg.h/ml because only free drug can go from the lung to the systemic circulation.On the other hand,administering a 20 mg oral dose may result in a systemic fl uticasone AUC of 1230 pg.h/ml but only 1%of this is free drug concentration,so a free AUC of only 12.3 pg.h/ml.Therefore an exposure of only 12.3 pg.h/ml can reach the lungs in terms of AUC.It is because only free drug concentrations are active and equilibrate through membranes resulting in a tiny exposure of fl uticasone in the lung(e.g.,AUC of 12.3 pg.h/ml)that a 20 mg fl uticasone oral dose was not ef fi cacious in that study,not that fl uticasone is not active from the systemic circulation as suggested[56].Considering that the fl uticasone AUC in the lung would only be 12.3 pg.h/ml with the 20 mg oral dose,it is not surprising that this dosing regimen was not ef fi cacious as the resulting AUC at the site of activity would be 10 times lower than what would be achieved with the lowest fl uticasone inhalation dosing regimen of 100 mcg BID.
For other ICS products that have higher gut bioavailability or low protein binding,systemic concentrations from gut absorption do present ef fi cacy and this absorption from other sites should not be ignored.For example,oral albuterol has an oral bioavailability of 44 to 63%[30,58]and negligible plasma protein binding at approximately 8%[59].Orally administered albuterol has been demonstrated to be ef fi cacious in asthma patients[60]with improved morning and evening FEV1values comparable to inhaled salmeterol indicating that albuterol administered orally was able to have an effect on the lung.
The importance of protein binding on the ef fi cacy of inhaled drugs has been recognized by Wu et al.where the authors showed that glucocorticoids with higher plasma and tissue binding showed a reduced ef fi cacy in the lung[61].
5. Future directions
5.1. Global harmonization and the pursuit of the global worldwide reference product
We have seen that there is still more work to do for highly regulated regions like the USA,Europe,and Canada to present and enforce aligned regulations regarding OIPs.In this current global economy,it would be very advantageous and less confusing if regulatory bodies of highly regulated regions such as FDA, EMA,and HC would present aligned regulations.There is a lot of“gray”in scienti fi c understanding(e.g.,it is not“black or white”)and regulations do follow science,so it is not surprising that opinions between regulators vary based on this level of“gray”.It may be reasonable scienti fi cally for EMA to suggest performing PK equivalence studies with charcoal and without charcoal,but then it may be reasonable for HC and FDA not to request it as well.The same can be said forTE studies.Regulators should grasp the opportunity to better align their regulations and guidances based on an understanding of what may be a common reasonable scienti fi c position.
Even more limiting,though,is the fact that FDA,EMA,and HC have to ask generic pharmaceutical companies to assess BE of their test product versus a reference product that is available in their own respective market.So theoretically even though regulations and guidances would be perfectly aligned, generic pharmaceutical companies would still need to perform all studies versus the US reference product,versus an EU reference product,and again versus the Canadian reference product.This is obviously not fi nancially viable nor desirable in regards to OIPs.Regulatory agencies do not share between themselves submitted pharmaceutical company dossiers,preventing them to assess if the reference product from one country is identical to the one in theirs.This has been the major impediment toward declaring a“global worldwide reference product”that generic pharmaceutical companies could use in their development.It is however very likely that most of the reference products marketed in highly regulated regions such as the USA,Europe,and Canada are in fact the identical products,making the topic even more frustrating to not just generic pharmaceutical companies themselves,but also to patients who need to pay more for their generic drugs because of greater development costs,and to regulatory agencies themselves who would save time and money if they could share between themselves the review of these submissions.We would propose that highly regulated regions such as the USA,Europe,and Canada agree to accept studies with either the US,EU,or Canadian reference products if all of the different product labels of these three reference products are quoting the exact same pivotal Phase III studies to establish their safety and ef fi cacy.When this is the case,it then means that all of these marketed reference products had to be proven to be bioequivalent to the formulation used in the pivotal Phase III trials,and therefore they should by extension be bioequivalent between each other.
Harmonizing regulations and accepting more often studies performed with a“foreign”reference product,or declaring a“global worldwide reference product”in appropriate cases, would bring more clarity to pharmaceutical companies,patients,and health care professionals as to what is required,and encourage more companies to invest in the development of products.
5.2. Drop of the requirement for TE studies if PK study is successful
We have seen that the usefulness of TE studies is limited on its own because of its very low discriminatory power when equivalence is assessed on the response scale,or because of its impossibility at assessing equivalence with the dosescale approach when the commercialized doses of the OIPs are already all associated with effects residing at the top of the response curve.TE studies for OIPs are prohibitively expensive.A single TE study that follows the FDA guidance for a generic formulation of Advair Diskus®,for example,may have to enroll thousands of patients at an overall cost listed in the 7 fi gures.If this study was absolutely necessary to establish clinical equivalence then it would be reasonable to perform. But understanding that this study is not really discriminative even if powered correctly,understanding that the systemic exposure PK equivalence study is on the other hand discriminative,and understanding that due to the high perfusion nature of the lung the systemic PK exposure should re fl ect the free drug PK exposure“in solution”at the site of ef fi cacy in the lung, then the conclusion is that TE studies should not be necessary to establish BE once device,in vitro,and PK equivalence are all established.
6. Conclusion
While there are many factors to consider with OIPs for local action in comparison with orally administered drugs,it can be argued that PK equivalence studies could be used to establish BE between such products without necessitatingTE studies with CE.Although some have argued that systemic drug concentrations that are used in PK equivalence assessments do not re fl ect drug levels or PK processes in the lungs,this is often incorrectly based on total drug instead of free drug concentrations.Because of the high-perfusion nature of the lungs, systemic exposure levels should be in equilibrium with and adequately inform on what would be the free active drug in solution at the site of activity in the lungs for OIPs.
REFERENCES
[1]Lipworth BJ.Pharmacokinetics of inhaled drugs.Br J Clin Pharmacol 1996;42(6):697-705.
[2]Rubin BK.Air and soul:the science and application of aerosol therapy.Respir Care 2010;55(7):911-921.
[3]Masoli M,Fabian D,Holt S,et al.Global burden of asthmadeveloped for the global initiative for asthma,<http://www .ginasthma.org/local/uploads/ fi les/GINABurdenReport_1 .pdf>;2015[accessed 28.06.15].
[4]Masoli M,Fabian D,Holt S,et al.Global initiative for asthma (GINA)-Global strategy for asthma management and prevention,<http://www.ginasthma.org/local/uploads/ if les/GINA_Report_2015_May19.pdf>;2015[accessed 28.06.15].
[5]Juniper EF,Kline PA,Vanzieleghem MA,et al.Effect of longterm treatment with an inhaled corticosteroid(budesonide) on airway hyperresponsiveness and clinical asthma in nonsteroid-dependent asthmatics.Am Rev Respir Dis 1990;142(4):832-836.
[6]The Childhood Asthma Management Program Research Group.Long-term effects of budesonide or nedocromil in children with asthma.N Engl J Med 2000;343(15):1054-1063.
[7]Jeffery PK,Godfrey RW,Adelroth E,et al.Effects of treatment on airway in fl ammation and thickening of basement membrane reticular collagen in asthma.A quantitative light and electron microscopic study.Am Rev Respir Dis 1992;145(4 Pt 1):890-899.
[8]Suissa S,Ernst P,Benayoun S,et al.Low-dose inhaled corticosteroids and the prevention of death from asthma. N Engl J Med 2000;343(5):332-336.
[9]Hizawa N.LAMA/LABA vs ICS/LABA in the treatment of COPD in Japan based on the disease phenotypes.Int J Chron Obstruct Pulmon Dis 2015;10:1093-1102.
[10]Global Initiative for Chronic Obstructive Lung Disease (GOLD).Global strategy for the diagnosis,management,and prevention of chronic obstructive pulmonary disease,<http://www.goldcopd.org/uploads/users/ fi les/GOLD _Report_2015.pdf>;2015[accessed 30.06.15].
[11]Aitken M,Berndt ER,Cutler D.Prescription drug spending trends in the United States:looking beyond the turning point.Health Aff 2009;28(1):w151-w160.
[12]US Food and Drug Administration.Facts about generic drugs-2015.<http://www.fda.gov/drugs/ resourcesforyou/consumers/buyingusingmedicinesafely/ understandinggenericdrugs/ucm167991.htm>;[accessed 16.08.15].
[13]The Canadian Generic Pharmaceutical Association(CGPA). The Canadian generic market year,<http://www .canadiangenerics.ca/en/resources/market_trends.asp>; 2013[accessed 26.06.15].
[14]US Food and Drug Administration.Challenges and opportunities report-March 2004.<http://www.fda.gov/ ScienceResearch/SpecialTopics/CriticalPathInitiative/ CriticalPathOpportunitiesReports/ucm077262.htm#f5>; [accessed 08.07.15].
[15]US Food and Drug Administration.Guidance for industry. Bioavailability and bioequivalence studies for orally administered drug products-general considerations,<http://www .fda.gov/downloads/Drugs/DevelopmentApprovalProcess/ HowDrugsareDevelopedandApproved/ApprovalApplications/ AbbreviatedNewDrugApplicationANDAGenerics/ UCM154838.pdf>;2002[accessed 01.07.15].
[16]US Food and Drug Administration.Guidance for industry. Metered dose inhaler(MDI)and dry powder inhaler(DPI) drug products.Chemistry,manufacturing,and controls documentation,<http://www.fda.gov/downloads/Drugs/ Guidances/ucm070573.pdf>;1998[accessed 08.07.15].
[17]US Food and Drug Administration.Draft guidance on budesonide,<http://www.fda.gov/downloads/Drugs/ GuidanceComplianceRegulatoryInformation/Guidances/ UCM319977.pdf>;2012[accessed 08.07.15].
[18]US Food and Drug Administration.Draft guidance on albuterol sulfate,<http://www.fda.gov/downloads/drugs/ guidancecomplianceregulatoryinformation/guidances/ ucm346985.pdf>;2013[accessed 05.07.15].
[19]US Food and Drug Administration.Draft guidance on fl uticasone propionate;salmeterol xinafoate,<http://www.fda.gov/downloads/Drugs/ GuidanceComplianceRegulatoryInformation/Guidances/ UCM367643.pdf>;2013[accessed 08.07.15].
[20]Lee S.Scienti fi c and regulatory considerations for bioequivalence(BE)of dry powder inhalers(DPIs). US Food and Drug Administration,Of fi ce of Generic Drugs,<http://www.fda.gov/downloads/ Drugs/DevelopmentApprovalProcess/ HowDrugsareDevelopedandApproved/ApprovalApplications/ AbbreviatedNewDrugApplicationANDAGenerics/UCM292652 .pdf>;2011[accessed 08.07.15].
[21]Lee SL,Saluja B,García-Arieta A,et al.Regulatory considerations for approval of generic inhalation drug products in the US,EU,Brazil,China,and India.AAPS J 2015;17(5):1285-1304.doi:10.1208/s12248-015-9787-8.
[22]Hochhaus G,Davis-Cutting C,Oliver M,et al.Current scienti fi c and regulatory approaches for development of orally inhaled and nasal drug products:overview of the IPAC-RS/University of Florida Orlando Inhalation Conference.AAPS J 2015;17(5):1305-1311.doi:10.1208/s12248 -015-9791-z.
[23]Health Canada.Guidance to establish equivalence or relative potency of safety and ef fi cacy of a second entry short-acting beta2-agonist metered dose inhaler,<http://www.hc-sc.gc .ca/dhp-mps/alt_formats/hpfb-dgpsa/pdf/prodpharma/ mdi_bad-eng.pdf>;1999[accessed 08.07.15].
[24]Health Canada.Guidance for industry-pharmaceutical quality of inhalation and nasal products,<http://www .hc-sc.gc.ca/dhp-mps/alt_formats/hpfb-dgpsa/pdf/ prodpharma/inhalationnas-eng.pdf>;2006[accessed 08.07.15].
[25]Health Canada.Draft guidance document-data requirements for safety and effectiveness of subsequent market entry inhaled corticosteroid products for use in the treatment of asthma for industry,<http://www.hc-sc.gc.ca/ dhp-mps/alt_formats/pdf/consultation/drug-medic/draft _inhal_ebauche_corticost-eng.pdf>;2011[accessed 08.07.15].
[26]European Medicines Agency.Guideline on the requirements for clinical documentation for orally inhaled products(OIP) including the requirements for demonstration of therapeutic equivalence between two inhaled products for use in the treatment of asthma and chronic obstructive pulmonary disease(COPD)in adults for use in the treatment of asthma in children and adolescents,<http://www.ema .europa.eu/docs/en_GB/document_library/Scienti fi c _guideline/2009/09/WC500003504.pdf>;2009[accessed 08.07.15].
[27]European Medicines Agency.Guideline on the pharmaceutical quality of inhalation and nasal products,<http://www.ema.europa.eu/docs/en_GB/document_library/ Scienti fi c_guideline/2009/09/WC500003568.pdf>;2006 [accessed 08.07.15].
[28]US Food and Drug Administration.Product label for Flovent Diskus( fl uticasone propionate inhalation powder 50,100, and 250 mcg),GlaxoSmithKline,<http://www.accessdata .fda.gov/drugsatfda_docs/label/2014/020833s028lbl.pdf>; 2014[accessed 09.07.15].
[29]Auclair B,Wainer IW,Fried K,et al.A population analysis of nebulized(R)-Albuterol in dogs using a novel mixed gut-lung absorption PK-PD model.Pharm Res 2000;17(10):1228-1235.
[30]Goldstein DA,Tan YK,Soldin SJ.Pharmacokinetics and absolute bioavailability of salbutamol in healthy adult volunteers.Eur J Clin Pharmacol 1987;32(6):631-634.
[31]Du XL,Zhu Z,Fu Q,et al.Pharmacokinetics and relative bioavailability of salbutamol metered-dose inhaler in healthy volunteers.Acta Pharmacol Sin 2002;23(7):663-666.
[32]Mager DE,Jusko WJ.Development of translational pharmacokinetic-pharmacodynamic models.Clin Pharmacol Ther 2008;83:909-912.
[33]Seng Yue C,Ducharme MP,D’Angelo P.Pharmacodynamics. In:Cartwright AC,Matthews BR,editors.International pharmaceutical product registration.New York:Informa Healthcare USA,Inc.;2009.
[34]US Food and Drug Administration.Draft guidance on orlistat,<http://www.fda.gov/downloads/drugs/ guidancecomplianceregulatoryinformation/guidances/ ucm201268.pdf>;2010[accessed 08.07.15].
[35]Barnes PJ,Adcock IM.How do corticosteroids work in asthma?Ann Intern Med 2003;139:359-370.
[36]O’Connor D,Adams WP,Chen ML,et al.Role of pharmacokinetics in establishing bioequivalence for orally inhaled drug products:workshop summary report.J Aerosol Med Pulm Drug Deliv 2011;24(3):119-135.
[37]Vijverberg SJ,Koenderman L,Koster ES,et al.Biomarkers of therapy responsiveness in asthma:pitfalls and promises. Clin Exp Allergy 2011;41(5):615-629.
[38]American Thoracic Society(ATS),European Respiratory Society(ERS).ATS/ERS recommendations for standardized procedures for the online and of fl ine measurement of exhaled lower respiratory nitric oxide and nasal nitric oxide, 2005.Am J Respir Crit Care Med 2005;171(8):912-930.
[39]Hendeles L,Daley-Yates PT,Hermann R,et al. Pharmacodynamic studies to demonstrate bioequivalence of oral inhalation products.AAPS J 2015;17(3):758-768.
[40]Lee SL.Investigation of dose-response of exhaled nitric oxide following oral inhalation of fl uticasone propionate.2012.Presented at 2012 GPhA/FDA Fall Technical Conference; Oct 02,2012.
[41]Gelb AF,Moridzadeh R,Singh DH,et al.In moderate-tosevere asthma patients monitoring exhaled nitric oxide during exacerbation is not a good predictor of spirometric response to oral corticosteroid.J Allergy Clin Immunol 2012;129:1491-1498.
[42]Barnes PJ,Dweik RA,Gelb AF,et al.Exhaled nitric oxide in pulmonary diseases.Chest 2010;138(3):682-692.
[43]Green RH,Brightling CE,Woltmann G,et al.Analysis of induced sputum in adults with asthma:identi fi cation of subgroup with isolated sputum neutrophilia and poor response to inhaled corticosteroids.Thorax 2002;57:875-879.
[44]Sze fl er SJ,Martin RJ,King TS,et al.Signi fi cant variability in response to inhaled corticosteroids for persistent asthma. J Allergy Clin Immunol 2002;109:410-418.
[45]Berry M,Morgan A,Shaw DE,et al.Pathological features and inhaled corticosteroid response of eosinophilic and noneosinophilic asthma.Thorax 2007;62:1043-1049.
[46]Pavord ID,Brightling CE,Woltmann G,et al.Noneosinophilic corticosteroid unresponsive asthma.Lancet 1999;353:2213-2214.
[47]Meijer RJ,Postma DS,Kauffman HF,et al.Accuracy of eosinophils and eosinophil cationic protein to predict steroid improvement in asthma.Clin Exp Allergy 2002;32:1096-1103.
[48]Godon P,Boulet LP,Malo JL,et al.Assessment and evaluation of symptomatic steroid-naive asthmatics without sputum eosinophilia and their response to inhaled corticosteroids.Eur Respir J 2002;20:1364-1369.
[49]Little SA,Chalmers GW,MacLeod KJ,et al.Non-invasive markers of airway in fl ammation as predictors of oral steroid responsiveness in asthma.Thorax 2000;55:232-234.
[50]Lex C,Jenkins G,Wilson NM,et al.Does sputum eosinophilia predict the response to systemic corticosteroids in children with dif fi cult asthma?Pediatr Pulmonol 2007;42: 298-303.
[51]Martin RJ,Sze fl er SJ,King TS,et al.Predicting response to inhaled corticosteroid ef fi cacy(PRICE)trial.J Allergy Clin Immunol 2007;119:410-418.
[52]Ducharme MP.Dose-scale pharmacodynamic bioequivalence studies-practical considerations.Presented at the AAPS Annual Meeting,San Diego,CA,USA Nov 6, 2014.
[53]Daley-Yates PT,Parkins DA.Establishing bioequivalence for inhaled drugs;weighing the evidence.Expert Opin Drug Deliv 2011;8(10):1297-1308.
[54]Persson G,Ankerst J,Gillen M,et al.Relative systemic availability of budesonide in patients with asthma after inhalation from two dry powder inhalers.Curr Med Res Opin 2008;24(5):1511-1517.
[55]Kerwin EM,Pearlman DS,de Guia T,et al.Evaluation of ef fi cacy and safety of budesonide delivered via two dry powder inhalers.Curr Med Res Opin 2008;24(5):1497-1510.
[56]Lawrence M,Wolfe J,Webb DR,et al.Ef fi cacy of inhaled fl uticasone propionate in asthma results from topical and not from systemic activity.Am J Respir Crit Care Med 1997;156(3 Pt 1):744-751.
[57]Patton JS,Fishburn CS,Weers JG.The lungs as a portal of entry for systemic drug delivery.Proc Am Thorac Soc 2004;1(4):338-344.
[58]Hindle M,Chrystyn H.Determination of the relative bioavailability of salbutamol to the lung following inhalation.Br J Clin Pharmacol 1992;34(4):311-315.
[59]Morgan DJ,Paull JD,Richmond BH,et al.Pharmacokinetics of intravenous and oral salbutamol and its sulphate conjugate.Br J Clin Pharmacol 1986;22(5):587-593.
[60]Martin RJ,Kraft M,Beaucher WN,et al.Comparative study of extended release albuterol sulfate and long-acting inhaled salmeterol xinafoate in the treatment of nocturnal asthma. Ann Allergy Asthma Immunol 1999;83(2):121-126.
[61]Wu K,Blomgren AL,Ekholm K,et al.Budesonide and ciclesonide:effect of tissue binding on pulmonary receptor binding.Drug Metab Dispos 2009;37(7):1421-1426.
(http://creativecommons.org/licenses/by-nc-nd/4.0/).
*Corresponding author.Learn and Con fi rm Inc.,3630 Bois Franc,St-Laurent,QC H4R 3K9,Canada.Tel.:+1 514 373 5346;fax:+1 514 373 5346. E-mail address:murray.ducharme@learnandcon fi rm.ca(M.P.Ducharme). Peer review under responsibility of Shenyang Pharmaceutical University.
http://dx.doi.org/10.1016/j.ajps.2015.08.006
1818-0876/©2015 The Authors.Production and hosting by Elsevier B.V.on behalf of Shenyang Pharmaceutical University.This is an open access article under the CC BY-NC-ND license(http://creativecommons.org/licenses/by-nc-nd/4.0/).
©2015 The Authors.Production and hosting by Elsevier B.V.on behalf of Shenyang Pharmaceutical University.This is an open access article under the CC BY-NC-ND license
 Asian Journal of Pharmacentical Sciences2015年6期
Asian Journal of Pharmacentical Sciences2015年6期
- Asian Journal of Pharmacentical Sciences的其它文章
- Inhaled nicotine replacement therapy
- Inhalation of nanoparticle-based drug for lung cancer treatment:Advantages and challenges
- Practical,regulatory and clinical considerations for development of inhalation drug products
- Mathematical approach for understandingdeagglomeration behaviour of drug powder in formulations with coarse carrier
- The effects of surface morphology on the aerosol performance of spray-dried particles within HFA 134a based metered dose formulations
- Delivery of theophylline as dry powder for inhalation