
Aln(n ﹤10000)团簇熔化行为的分子动力学模拟
2015-07-13 03:39伊力亚尔海米提李春丽段海明
原子与分子物理学报 2015年1期
伊力亚尔·海米提,李春丽,方 萌,段海明
(新疆大学物理科学与技术学院,乌鲁木齐830046)
1 引 言
团簇,作为几个乃至几千万个原子、离子或分子的聚集体,具有独特的物理化学性质[1],在新材料、纳米电子器件和纳米催化剂等领域有重要的应用价值,已引起研究者们的广泛关注[2-7].对团簇的形成、结构、性质及其尺寸演变过程的研究已经成为一个新的重要领域.
团簇的熔化行为明显不同于块体(晶体)熔化,其熔点通常随着尺寸(团簇所含原子数)的减小而降低,并且伴随有明显的预熔化温度区间,在尺寸较小时还体现出明显的尺寸效应(熔点随尺寸非单调变化)及奇特的熔化行为(如固液共存态). 金属团簇的熔化行为是人们尤为关注的性质之一. 如,Jarrold 小组研究发现熔化可以使铝团簇的活性增强[8];Mottet 等人发现通过掺杂可以改变银团簇的熔点[9];我们此前也探讨了Co团簇(Co14及Co56)的表面熔化现象[10]. 此外,对金属团簇熔化过程中的结构演化行为的研究也受到关注. 如,Schebarchov 等人在研究中发现镍团簇在固液共存时在一定条件下团簇结构由正二十面体结构转变为十面体结构[11];Wen 等详细研究了二十四面体结构的铂团簇在熔化过程中的结构演化[12];Xiao 等人研究发现Oh结构的Ag309团簇在存在错位等缺陷时Oh结构向Ih结构的转化温度升高[13].
三价金属铝晶体具有优良的导电导热性能和优异的延展性,已经在社会生产实践当中获得了广泛的应用. 对铝团簇的研究也揭示出其一系列奇特性质,如极小尺寸时铝团簇的铁磁性[14]、的超稳定性及其作为未来团簇组装材料的潜力[15]及优良的催化性能[16]等. 近年来,Jarrold小组对于中小尺寸Aln(n <200)团簇的熔化行为进行了大量的实验研究,发现了一系列奇特的熔化行为:如铝团簇的活性增强[8],小尺寸(n<100)时铝团簇熔点随尺寸变化的振荡现象,较大尺寸(n >100)时铝团簇比热呈现双峰的熔化现象等[17,18]. 之前我们对中小尺寸Aln(n <200)团簇的熔化行为进行了分子动力学模拟研究,得出了与实验一致的结论(如小尺寸铝团簇熔点与尺寸的反常依赖关系及中小尺寸铝团簇熔化的比热双峰现象)[19,20].
目前对于金属团簇熔化行为的研究主要集中于中小尺寸,而对较大尺寸金属团簇熔化行为的系统研究仍比较少见. 本文即采用分子动力学模拟方法结合退火技术,基于半经验的Gupta 多体势,从高对称性(Ih、Oh)密堆积满壳结构及退火结构出发,系统研究了包含10000 原子之内的铝团簇的熔化行为.
2. 势模型和计算方法
2.1 团簇初始结构
研究团簇的熔化行为,首先要确定团簇的初始结构. 一般是从某一低能结构(如基态结构)出发,研究团簇的升温(熔化)行为. 通常初始结构可由模拟退火算法求得,但在团簇尺寸较大时,模拟退火算法找到团簇最低能量(基态)结构的效率很低. 本文在对若干Aln(n <10000)团簇熔化行为的分子动力学模拟中的初始结构的确定,除考虑退火算法所得结构外,还考虑了两种常见的密堆积高对称性结构:Ih(Icosahedron)结构和Oh(Truncated Octahedron)结构. 由于研究尺寸范围较大,所以我们只对满足上述两类对称性的满壳层结构尺寸进行了模拟研究.
满壳层Ih结构团簇所含原子数n 与壳层数k的关系可表示为[21]:
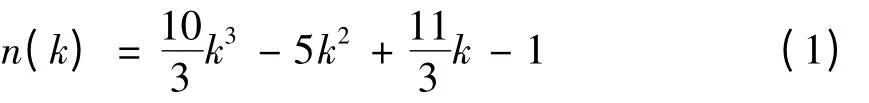
本文研究满壳层Oh结构可以通过截断正八面体的六个顶点得到. 当正八面体边长nl和截断距离ncut满足关系式nl=2ncut+1 时,满壳层Oh结构团簇所含原子数n 与Ih结构相同. 在本文研究Aln团簇尺寸范围内(n <10000),所模拟满壳层Ih及Oh结构团簇尺寸序列为:13、55、147、309、 561、 923、 1415、 2057、 2869、 3871、5083、6525、8217,共计13 个不同尺寸.
2.2 势函数
本文研究Al 团簇其原子间相互作用势采用半经验的Gupta 多体势函数描述. Gupta 势是基于紧束缚模型二阶矩近似的嵌入原子势,这种多体势已被广泛应用于研究金属及合金金属团簇的几何结构[22,23]及动力学行为[24]. Gupta 势可由原子间Born -Mayer 形式的相互作用排斥项和含多体效应的吸引项组成,形式如下
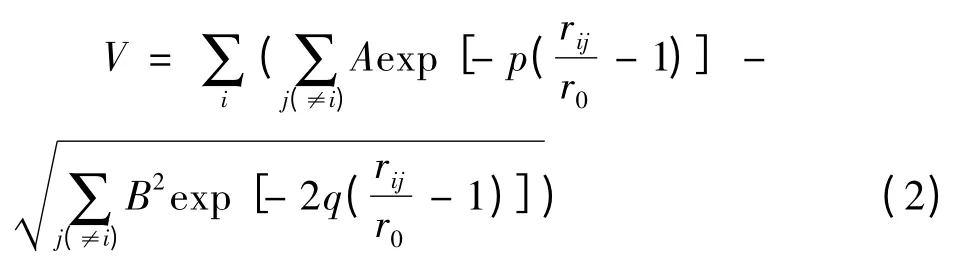
其中rij为i 、j 原子间距. r0是该原子的特征长度(文中选为Al 晶体最近邻原子间距),A 是衡量原子间排斥强度的量,B 是有效跳跃积分、通常只与原子类别有关. 本文研究Al 团簇其Gupta 势参数为A =0.1221 eV、B =1.316 eV、p =8.612、q=2.516 及r0=2.86. 这些参数是通过拟合块体Al材料的束缚能、晶格常数、体弹模量及弹性扭转常数而得到的[25].
2.3 分子动力学
本文采用恒温分子动力学方法,温度由Berendsen 热浴标度[26],采用速度Verlet 算法积分牛顿运动方程. 体系从初始结构出发(如上Ih及Oh满壳层结构,及退火结构,详见下述),积分牛顿方程时间步长为1fs,每个温度点进行5 ×106步MD 模拟. 模拟温度范围取为200K 到1200K,温度间隔选为20K.
除考虑如上两种高对称性(Ih及Oh)满壳层密堆积结构外,还采用了分子动力学模拟退火方法获取团簇的初始结构. 由退火方法获得团簇初始稳定结构的步骤如下:对任一团簇初始稳定结构(如可由随机数结合最速下降法得到)直接升温至1200K (确保团簇处于类液态),再进行缓慢降温的分子动力学退火过程,降温直至200K(确保团簇处于类固态),降温过程温度间隔取为20K,再对200K 时最终结构作最速下降法弛豫至稳定结构(称为退火结构).
2.4 计算物理量
对如上每一次分子动力学模拟,在每个温度点上计算体系的比热. 体系比热按下式计算:
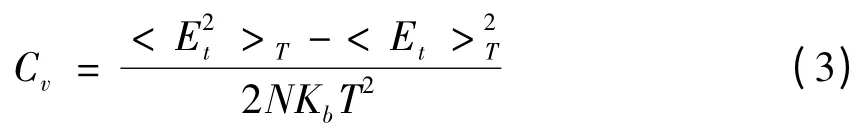
其中Kb为Boltzmann 常数,Et为体系的总能,T为体系的温度, <>T为系综平均(即长时间平均).
3 结果分析与讨论
3.1 团簇初始结构
表1 给出13 个不同尺寸时Aln(n = 13 ~8217)团簇三种不同初始(稳定)结构的总(结合)能量. 对比分析各能量可见:在所有尺寸下Ih结构能量均为最低,Oh结构能量均高于Ih结构,考虑到铝晶体为Oh(FCC)结构,这也说明Al 团簇在包含接近10000 个原子时其几何结构还没有过渡到Al 晶体,但两类结构平均每原子结合能差值却在减小,如从Al13的约0.0531 (eV/atom)减小到Al8217的约0.0009 (eV/atom);此外,从表1 中退火结构的能量可大致分析退火方法寻找团簇基态结构的效率问题,在团簇尺寸较小时(如309 个原子之内),退火所得的结构就是Ih结构. 然而随团簇尺寸增加,所得退火结构的能量均高于Ih结构,而且随着尺寸的变大其差距也变大,表明退火结构找基态结构的效率逐渐变低,当团簇尺寸大到3871 时,退火结构的能量甚至高于Oh结构.
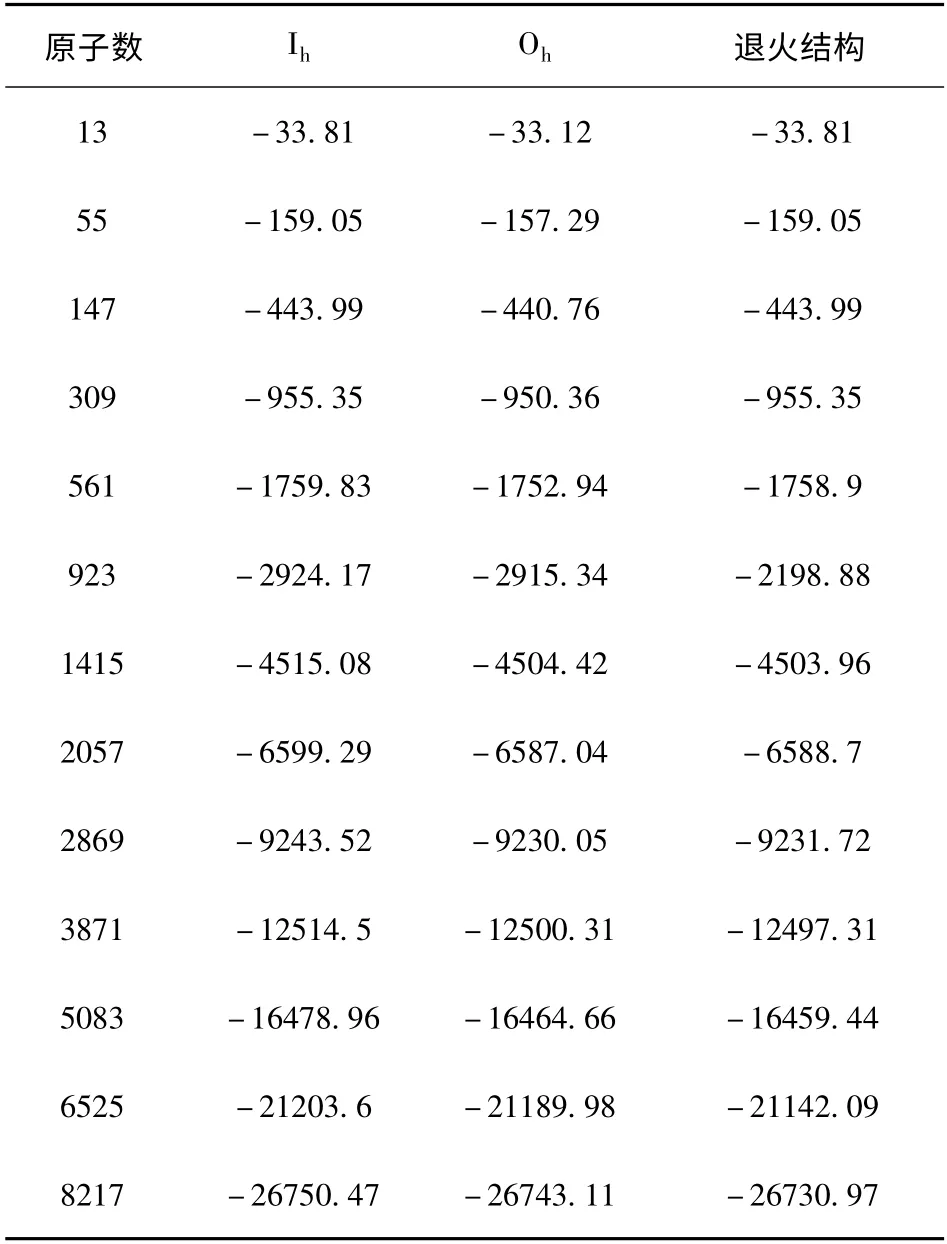
表1 不同尺寸时Aln (n=13 ~8217)团簇三种结构的能量(eV)Table 1 Energies (in eV)of three different structures of Aln(n=13 ~8217)clusters at different sizes
3.2 团簇的熔化行为及稳定性
图1 (a)、(b)及(c)分别给出从三种不同初始结构(Ih、Oh及退火(Annealing)结构)出发模拟所得不同尺寸Aln(n = 309、561 及923)团簇熔化过程中的能量(E,左图)和比热(Cv,右图)随温度(T)的变化关系图.
对于Al309(图1 (a)),从能量随温度变化图可看出三种初始结构所对应的曲线几无分别.这是因为Oh结构在小尺寸(309 原子)时动力学极不稳定,在很低温度(模拟初始温度)下已经转变为动力学更加稳定的(类)Ih结构. 而Al309退火结构就是Ih结构(见表1 所示),所以三条曲线都可视为是从同一个结构出发所得,三个曲线几乎重叠. Al309的比热随温度变化图 (图1(a)右侧)出现了一个明锐的的单峰,其对应的温度点为660K,此即为Al309团簇的熔点.
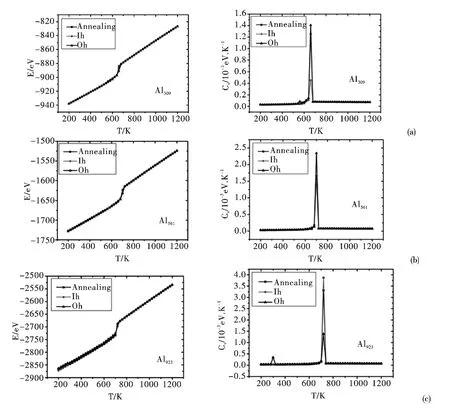
图1 Al309 (a)、Al561 (b)及Al923 (c)团簇的能量(E)和比热(Cv)随温度(T)的变化关系Fig.1 Energies (E)and the heat capacities (Cv)of Al309 (a),Al561 (b)and Al923 (c)clusters as a function of temperature (T)
对于Al561(图1 (b)),尽管其退火结构不是完整Ih结构(但也是类Ih结构,即相对于完整Ih结构只是部分原子有偏离),在模拟初始温度(200K)时,其结构也是类Ih结构,所以其曲线与Ih结构所对应曲线几乎重合. 而Oh初始结构对应曲线在模拟初始温度(200K)时结构已经转变为类Ih结构,其能量曲线随温度的变化关系也变得与Ih结构对应曲线基本相同(为探究Al561团簇的类Oh结构向类Ih结构转变发生时的温度区间,我们也从更低的初始温度(100K)出发模拟研究了Al561团簇的熔化行为,发现其结构转变发生在160K). Al561的比热曲线随温度变化图(图1 (b)右侧)出现了一个尖锐的峰,该峰值所对应的温度点(700K)即为Al561团簇的熔点.
由图1 (c)可见,Al923退火结构所对应的能量曲线与Ih结构对应曲线不同(前者能量略高).由前述(表1)可知,Al923退火结构并非Ih结构,而且能量明显高于Ih结构(平均每原子能量差也明显大于Al561团簇的Ih结构与退火结构的能量差). Ih结构对应曲线依然保持能量最低,而Oh结构在300K 时转变为Ih结构,这表明随着团簇尺寸变大,Oh结构动力学稳定性变强,所以向Ih结构的转变温度也随之升高. 在Al923比热随温度的变化图(图1 (c)右侧)中,300K 和720K时的两个尖锐的峰分别对应于类Oh到类Ih结构转变对应的温度点及Al923团簇的熔点.
图2 (a)及(b)分别给出从三种不同初始结构(Ih、Oh及退火(Annealing)结构)出发模拟所得不同尺寸Aln(n =2057 及8217)团簇熔化过程中的能量(E,左图)和比热(Cv,右图)随温度(T)的变化关系图. 从图2 (a)可以看出,直至团簇熔化Oh结构所对应的能量随温度变化曲线始终都是一条独立的曲线,这表明在完全熔化之前,Al2057始终保持了类Oh结构而没有转变为类Ih结构,说明Al2057团簇的(类)Oh结构在完全熔化前的全部温度范围内均为动力学稳定的. 在团簇尺寸n≥2057 时,(多次)模拟Aln熔化行为所得Oh结构所对应的能量随温度变化曲线在团簇完全熔化前都表现为独立曲线,这表明:对Al 团簇,在包含约2000 原子以上时,其(类)Oh结构(在完全熔化发生前)是动力学稳定的.
对于其它满壳层Aln团簇(n =2869 ~8217之间),从三种不同初始结构 (Ih、Oh及退火(Annealing)结构)出发模拟所得团簇熔化过程中的能量和比热随温度的变化关系与如上Al2057是类同的(如图2 (b)所示Al8217情形).
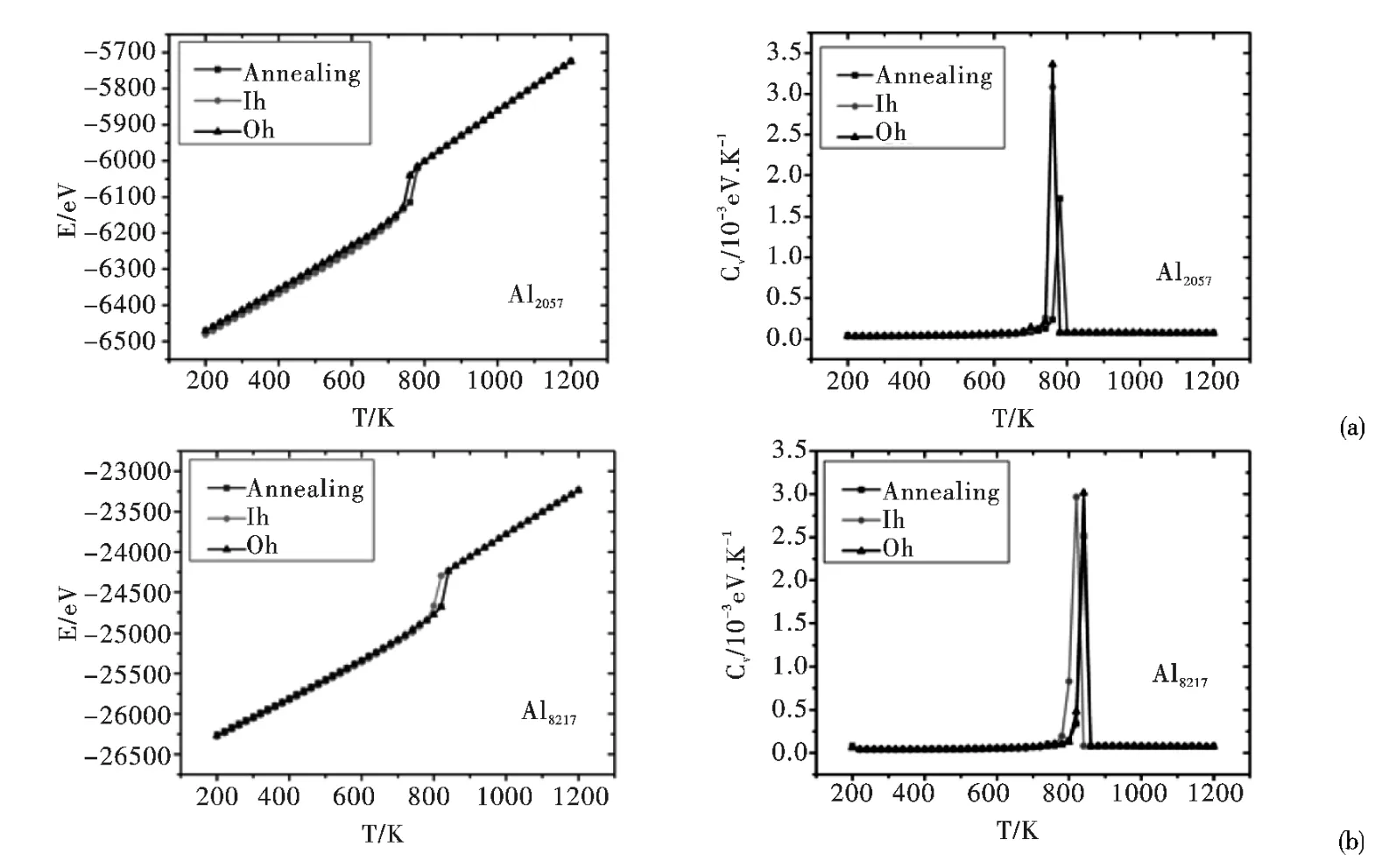
图2 Al2057 (a)及Al8217 (b)团簇的能量(E)和比热(Cv)随温度(T)的变化关系Fig.2 Energies (E)and the heat capacities (Cv)of Al2057 (a)and Al8217 (b)clusters as a function of temperature (T)
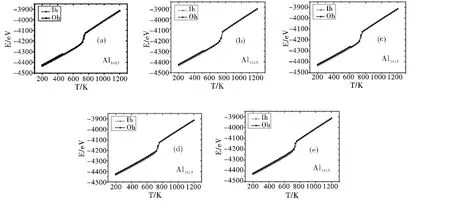
图3 五次不同模拟过程中Al1415团簇的能量(E)随温度(T)的变化关系Fig.3 Energies (E)of Al1415 cluster as a function of temperature (T)at five different simulations
如上所述,对于Oh结构高对称性满壳层Aln团簇:在所含原子数较少(n <1000)时,团簇动力学稳定性不高,在远离完全熔化时就会发生结构转变(由类Oh结构转变为类Ih结构);而在所含原子数较多(n >2000)时,团簇具有高的动力学稳定性,在完全熔化发生前均不会发生结构转变;则,对于尺寸介于1000 ~2000 之间的Aln团簇,其动力学稳定性又如何?图3 给出由Oh初始结构出发进行5 次不同模拟所得Al1415团簇熔化过程中能量随温度的变化关系,作为对比,同图中也给出了由Ih初始结构出发的相应曲线.由图3 可见,Al1415团簇的熔化行为是非常有趣的,5 次不同模拟所得熔化行为(结构演化)不尽相同,大致可以归为两类情形:(1)在完全熔化前发生了结构变化,由类Oh结构转化为类Ih结构(5 次不同模拟中有3 次属于此类情形,见图3 (a) ~ (c),此3 次模拟结果的区别仅在于结构变化发生时的温度不尽相同); (2)在完全熔化前未发生结构变化(对应于5 次不同模拟中的2 次结果,见图3 (d)、(e)). 我们认为,这恰好体现出Oh结构Aln团簇的动力学稳定性与团簇尺寸有密切关联:在较小尺寸(n <1000)时动力学不稳定,在较大尺寸(n >2000)时动力学稳定,而在中间尺寸(n =1000 ~2000)时,团簇动力学稳定性具有一定的随机性,体现出一个过渡区行为.
3.3 团簇熔点随团簇尺寸及初始结构变化
图4 中给出了本文所研究全部Aln(n =13 ~8217)团簇从三种不同初始结构出发所得比热曲线的峰值(团簇熔点)随团簇尺寸n-1/3的变化关系图,相应各熔点的具体数值列于表2 中. 由图可见:对Al55及更大尺寸团簇,团簇熔点随尺寸变化基本上呈现出单调变化趋势,而在包含约200 以上原子时(Al309及更大尺寸团簇),Al 团簇熔点与团簇尺寸的负三分之一次方呈现出近似的线性关系,该线性关系的外推值(与纵轴的交点,对应于Al 晶体情形)落在930K 附近,对应于Al 晶体的熔点(933K);由图4 还可以看出,Al13团簇具有很高的熔点(约860K),高于本文所研究其它所有满壳层尺寸Aln(n =55 ~8217)团簇的熔点. 关于Al13及其附近团簇(如Al14~Al17)的高熔点以及小尺寸Aln(n =13 ~32)团簇的震荡式的熔点尺寸变化关系,我们此前已进行了详细的研究,得到了与实验观测一致的结论[19].
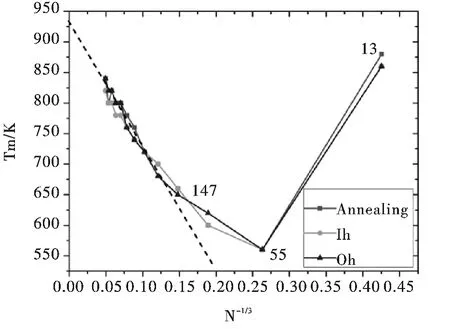
图4 Aln (n=13 ~8217)团簇的熔点随n -1/3 的变化图Fig.4 The melting points of Aln (n=13 ~8217)clusters as a function of n -1/3
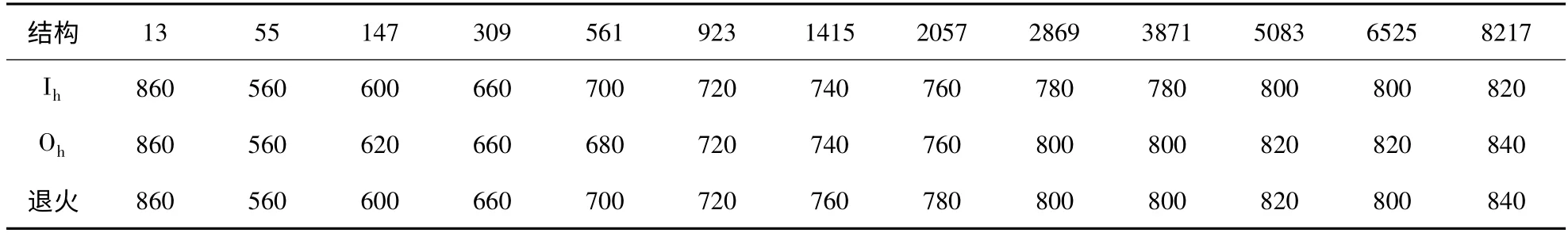
表2 由三种不同初始结构出发时模拟所得不同尺寸Aln (n=13 ~8217)团簇的熔点(K)Table 2 The melting points (in K)of Aln (n=13 ~8217)clusters at different sizes started from three different initial structures
需要说明的是,由表2 (图4)可以看出,对于相同尺寸Al 团簇,从不同初始结构出发时所得团簇熔点基本相同. 有些尺寸下从不同初始结构出发时的熔点虽然不相等,但是其差距没有超出模拟所选取的最小温度变化间隔(20K),对此我们也对如上不同尺寸不同初始结构各Al 团簇进行了多次模拟,所得结果(相同尺寸时熔点)差别均在20K 之内,所以我们认为,对于本文所研究Al 团簇,团簇初始结构对团簇熔点没有影响.
4 结 论
本文基于半经验的Gupta 原子间多体相互作用势函数,运用分子动力学方法并结合退火技术系统模拟研究了尺寸在10000 个原子之内的Al 团簇的熔化行为,分析讨论了团簇能量及比热随温度的变化关系. 对于各给定尺寸团簇,均考虑了三种不同的初始结构:高对称性密堆积满壳层的Ih、Oh结构及退火结构. 模拟结果表明:(1)在所研究团簇尺寸范围内,二十面体(Ih)结构均显示出高的动力学稳定性,在团簇完全熔化前无结构转变发生;(2)Oh结构的动力学稳定性与团簇尺寸(团簇所包含原子数)密切相关:在团簇尺寸较小(1000 个原子以内)时Oh结构不稳定(熔化前会发生向类Ih结构的转变);在中间尺寸(1000 ~2000 原子间)时Oh结构动力学稳定性发生转变,相应于一个过渡区域;在团簇尺寸较大(2000 原子以上)时Oh结构显示出完整的动力学稳定性; (3)对同一尺寸Al 团簇,从不同初始结构出发模拟所得团簇熔点基本相同;(4)在包含50 原子以上时,团簇熔点随尺寸变化基本上呈现出单调变化趋势,而在包含约200 以上原子时,Al 团簇熔点与团簇尺寸的负三分之一次方呈现出近似的线性关系.
[1] Wang G H. Cluster Physics[M]. Shanghai:Shanghai Scientific and Technology Press,2003(in Chinese)[王广厚. 团簇物理学[M]. 上海:上海科学技术出版社,2003]
[2] Baletto F,Ferrando R. Structural properties of nanoclusters: Energetic, thermodynamic, and kinetic effects[J]. Rev. Mod. Phys.,2005,77:371.
[3] Bergeron D E,Roach P J,Khanna S N. Al cluster superatoms as halogens in polyhalides and as alkaline earths in iodide salts[J]. Science,2005,307:231.
[4] Castleman A W,Jena P. Clusters:A bridge across the disciplines of environment,materials science,and biology[J]. PNAS,2006,103:10554.
[5] Meng K L,Zhang F S,Zhang Y P,et al. Effect of geometry structure on melting process of Na8cluster[J].J. At. Mol. Phys.,2006,23(2):353(in Chinese)[蒙克来,张丰收,张艳萍,等.几何结构对Na8团簇熔化过程的影响[J]. 原子与分子物理学报,2006,23(2):353]
[6] Wang Z G,Qiu S Y,Wen Y H. Molecular dynamics simulations of the melting of nickel nanoclusters[J].J. At. Mol. Phys.,2008,25(4):848(in Chinese)[汪志刚,邱姝颖,文玉华.纳米团簇熔化过程的分子动力学模拟[J]. 原子与分子物理学报,2008,25(4):848]
[7] Liu J T,Duan H M. Molecular dynamics simulation of structures and melting behaviours of iridium clusters with different potentials[J]. Acta Phys. Sin.,2009,58:4826(in Chinese)[刘建廷,段海明. 不同势下铱团簇结构和熔化行为的分子动力学模拟[J]. 物理学报,2009,58:4826]
[8] Cao B P,Starace A K,Jarrold F M,et al. Melting dramatically enhances the reactivity of Aluminum nanoclusters[J]. J. Am. Chem. Soc.,2009,131:7.
[9] Mottet C,Rossi G,Ferrando R,et al. Single impurity effect on the melting of nanoclusters[J]. Phys. Rev.Lett.,2005,95:035501.
[10] Lu S W,Zhang J,Duan H M. Melting behaviors of CoN(N = 13,14,38,55,56)clusters[J]. Chem.Phys.,2009,363:7.
[11] Schebarchov D,Hendy S C. Transition from icosahedral to decahedral structure in a coexisting solid -liquid nickel cluster[J]. Phys. Rev. Lett.,2005,95:116101.
[12] Wen Y H,Fang H,Sun S G,et al. A molecular dynamics study of shape transformation and melting of tetrahexahedral platinum nanoparticle [J]. Chem.Rev. Lett.,2009,471:295.
[13] Xiao X Y,Dai W C,Xia J H. The study of structural transformation for Ag309clusters with molecular dynamics method[J]. J. At. Mol. Phys.,2013,30(4):585(in Chinese)[肖绪洋,代武春,夏继红. Ag309团簇升温过程中结构转变的分子动力学模拟研究[J]. 原子与分子物理学报,2013,30(4):585]
[14] Cox D M,Trevor D J,Whetten R L,et al. Aluminum clusters:magnetic properties[J]. J. Chem. Phys.,1986,84:4561.
[15] Gong X G,Kumar V. Enhanced stability of magic clusters:A case study of icosahedral Al12X,X = B,Al,Ga,C,Si,Ge,Ti,As[J]. Phys. Rev. Lett.,1993,70:2078.
[16] Shimojo F,Ohmura S,Kalia R K,et al. Molecular dynamics simulations of rapid hydrogen production from water using aluminum clusters as catalyzers[J]. Phys.Rev. Lett.,2010,104:126102.
[17] Neal C M,Starace A K,Jarrold M F,et al. Melting of aluminum cluster cations with 31 -48 atoms:Experiment and theory[J]. J. Phys. Chem.,2007,111:17788.
[18] Starace A K,Cao B P,Judd O H,et al. Melting of size-selected aluminum nanoclusters with 84 -128 atoms[J]. J. Chem. Phys.,2010,132:34302.
[19] Li C L,Duan H M,Kerem M. Molecular dynamical simulation of the melting properties of Aln(n =13 -32)clusters [J]. Acta Phys. Sin.,2013,62:193104(in Chinese)[李春丽,段海明,买力坦·开来木. Aln(n=13 -32)团簇熔化行为的分子动力学模拟研究[J]. 物理学报,2013,62:193104][20] Li C L,Kerem M,Duan H M. Molecular dynamic simulation of the structural and melting properties of Al196cluster[J]. J. At. Mol. Phys.,2013,30(1):91 (in Chinese)[李春丽,买力坦·开来木,段海明. Al196团簇结构及熔化行为的分子动力学模拟[J]. 原子与分子物理学报,2013,30(1):91]
[21] Baletto F,Ferrando R,Fortunelli A,et al. Crossover among structural motifs in transition and noble -metal clusters[J]. J. Chem. Phys.,2002,116:3856.
[22] Michaelian K,Rendón N,Garzón I L. Structure and energetics of Ni,Ag,and Au nanoclusters[J]. Phys.Rev. B,1999,60:2000.
[23] Rodrí guez-López J L,Aguilera-Granja F,Michaelian K,et al. Structure and magnetism of cobalt clusters[J]. Phys. Rev. B,2003,67:174413.
[24] Zhang W,Zhang F S,Zhu Z Y. Molecular dynamics study on the melting phase transition of aluminum clusters with around 55 atoms[J]. Phys. Rev. B,2006,74:033412.
[25] Cleri F,Rosato V. Tight-binding potentials for transition metals and alloys[J]. Phys. Rev. B,1993,48:22.
[26] Berendsen H J C,Postma J P M,van Gunsteren W F,et al. Molecular dynamics with coupling to an external bath[J]. J. Chem. Phys.,1984,81:3684.
猜你喜欢
空气动力学学报(2022年4期)2022-08-23
黑龙江大学自然科学学报(2022年1期)2022-03-29
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
陶瓷学报(2021年1期)2021-04-13
江苏农业科学(2017年10期)2017-07-21
浙江大学学报(工学版)(2015年2期)2015-05-30
火炸药学报(2014年1期)2014-03-20
青年文摘·上半月(1982年2期)1982-01-01
