
外周全血中与老化相关的DNA甲基化标记的来源
2015-12-02 02:12李红东洪贵妮郭政
遗传 2015年2期
李红东,洪贵妮,郭政,2
外周全血中与老化相关的DNA甲基化标记的来源
李红东1,洪贵妮1,郭政1,2
1. 电子科技大学生命科学与技术学院,成都610054;2. 福建医科大学基础医学院,消化道恶性肿瘤教育部重点实验室,福州350108
机体老化与癌症、神经退行性疾病等许多复杂疾病相关。目前,研究者已在外周全血中识别了大量的与老化相关的DNA甲基化标记,这些标记可能反映外周血白细胞在机体老化过程中发生的变化,也可能反映外周血中与年龄相关的细胞构成比例的变化。文章利用3组正常个体外周全血DNA甲基化谱,采用Spearman秩相关分析识别了与老化相关的CpG甲基化位点(age-related DNA methylation CpG sites, arCpGs)并评价了其可重复性;利用去卷积算法估计了各外周血样本中髓性和淋巴性细胞的比例并分析了其与年龄的相关性;比较了在外周全血、CD4+T细胞和CD14+单核细胞中识别的arCpGs的一致性。结果显示,在独立外周全血数据中识别的arCpGs具有显著的可重复性(超几何检验,=1.65×10-11)。外周血髓性和淋巴性细胞的比例分别与年龄显著正、负相关(Spearman秩相关检验,<0.05,≤0.22),它们间DNA甲基化水平差异较大的CpG位点倾向于在外周全血中被识别为arCpGs。在CD4+T细胞中识别的arCpGs与在外周全血中识别的arCpGs显著交叠(超几何检验,=6.14×10-12),且99.1%的交叠位点在CD4+T细胞及外周全血中的DNA甲基化水平与年龄的正、负相关性一致。尽管在CD14+单核细胞中识别的arCpGs与在外周全血中识别的arCpGs并不显著交叠,但是在交叠的51个arCpGs中,有90.1%的位点在CD14+单核细胞、外周全血以及CD4+T细胞中的DNA甲基化水平与年龄的正、负相关性一致,提示它们可能主要反映细胞间共同的改变。在外周全血中识别的arCpGs主要反映某些白细胞共同或特异的DNA甲基化改变,但是也有一部分反映外周血细胞比例构成的变化。
外周全血;老化;DNA甲基化;CD4+T细胞;CD14+单核细胞
DNA甲基化作为一种重要的表观遗传修饰机制与机体老化密切相关[1,2]。在人类正常组织中,全基因组的DNA甲基化水平会随着年龄增长呈现整体去甲基化的现象,而在某些基因(如与细胞分化相关的梳状蛋白靶基因等)的特异CpG位点上会呈现高甲基化现象[3~5]。这些与老化相关的DNA甲基化改变能够增加基因组的不稳定性,改变基因表达水平,从而影响各组织的正常生物学功能,提高包括癌症在内的许多衰老相关疾病的发病风险[1]。在外周血中,人们已经观察到了与组织类似的DNA甲基化改变[6,7]。例如,通过比较百岁老人外周全血和婴儿脐胎血中的DNA甲基化模式,发现老人外周全血中的DNA甲基化模式整体上呈现去甲基化趋势,而与细胞分化相关的梳状蛋白靶基因却呈现高甲基化状态[6]。因此,许多研究者尝试在外周血中寻找年龄相关的DNA甲基化改变,并试图在组织中进行验证,以期找到可以代替组织分子标记的外周血分子标记[8,9]。
研究发现,外周血中与天然免疫和适应性免疫相关的细胞会随着年龄增大而发生功能变化[10]。例如,巨噬细胞和树突状细胞的噬菌作用、自然杀伤细胞的毒性、T细胞的增生能力会随着年龄增长而减弱[11,12]。外周血细胞的这些功能改变可能与DNA甲基化异常相关,例如T细胞随年龄发生的功能改变可能与T细胞内DNA整体去甲基化相关[13]。但是,目前尚不清楚在不同白细胞中随着老化发生的DNA甲基化改变是否相同以及这些改变能否在全血中得到反映。研究者还发现,外周血白细胞的构成比例也会随着年龄增大而发生变化,例如B细胞和CD8+T细胞的比例会随着年龄增大而减少[14]。由于来自髓性前体细胞和淋巴性前体细胞的白细胞(分别简称为髓性细胞和淋巴性细胞)之间的DNA甲基化模式存在较大差异[15,16],这种随年龄变化而产生的细胞构成比例的变化也可能对外周全血中老化相关的DNA甲基化信号产生影响[15]。因此,在外周全血中识别与老化相关DNA甲基化改变时,有必要分析其来源。
本文利用3组正常个体的外周全血DNA甲基化谱,首先证明在独立数据中识别的与老化相关的DNA甲基化CpG位点(age-related DNA methylation CpG sites, arCpGs)具有显著的可重复性。然后,利用去卷积算法估计了各外周全血样本中髓性和淋巴性细胞的比例,并分析了这两类细胞的比例与年龄的相关性及其对外周全血中识别的arCpGs的影响。最后,利用淋巴性细胞亚型CD4+T细胞和髓性细胞亚型CD14+单核细胞的甲基化谱数据,分析了不同白细胞亚型的arCpGs与在外周全血中识别的arCpGs的关系。
1 材料和方法
1.1 数据
本文所分析的DNA甲基化谱数据集均来自GEO(Gene Expression Omnibus)数据库[17]。各数据集的详细信息见表1。数据集Set1、Set2与Set3是同一个GEO数据系列(GEO检索号为GSE41037)中的3组独立数据集,分别包含193、105和96个正常的荷兰人外周全血DNA甲基化谱[8]。这3组数据集用来寻找正常人群外周全血中的arCpGs。数据集Set4包含正常外周血中NK细胞、B细胞、T细胞、单核细胞和粒细胞的DNA甲基化谱[18]。该数据集用作去卷积算法的参考数据集。数据集Set5和Set6分别包含来自正常个体外周血CD4+T细胞和CD14+单核细胞的DNA甲基化谱,这两组数据集分别用于寻找CD4+T细胞和CD14+单核细胞中的arCpGs[19]。
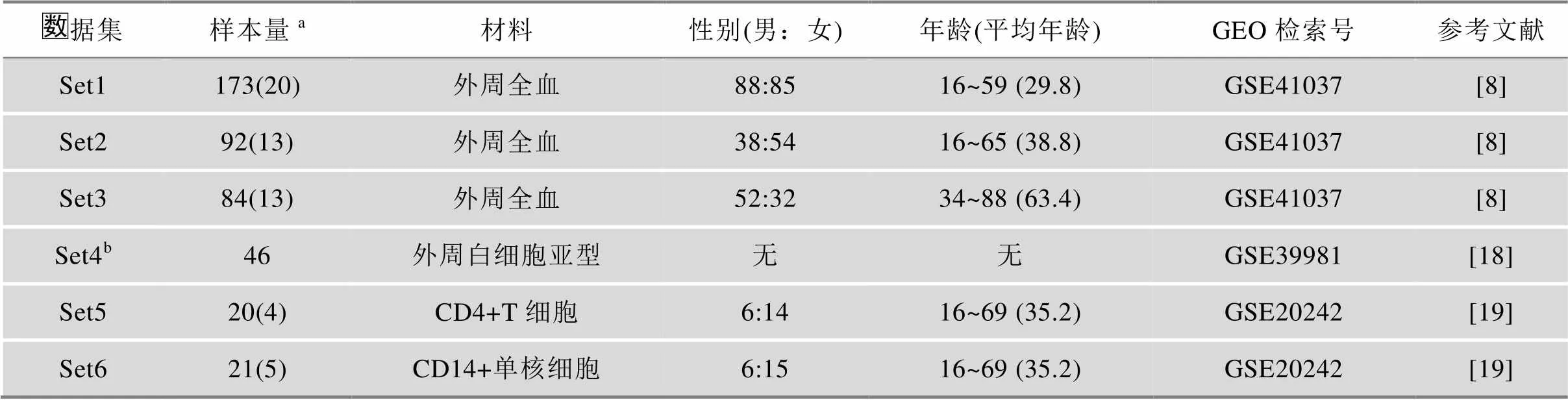
表1 DNA甲基化谱数据集
注:a括号外的数字是经过数据质量控制后保留下来用作后续分析的样本,括号内的数字是经数据质量控制后去掉的样本数。bSet4包含的外周白细胞亚型有NK细胞(n=12)、B细胞(n=5)、T细胞(n=16)、单核细胞(n=5)和粒细胞(n=8)。
1.2 数据预处理
本文所分析的DNA甲基化谱数据集均由Illumina Infinium HumanMethylation27 BeadChip平台检测。该平台共检测了14 495个基因启动子和转录起始位点附近的27 578个CpG位点的甲基化水平,本文只分析常染色体上的26 486个CpG位点。对于每个CpG位点,其DNA甲基化水平()由该位点的甲基化信号值()和非甲基化信号值()通过公式(1)计算[20]:

由于DNA甲基化水平检测容易受外界因素的影响,本文采用如下的步骤来控制每组数据集的质量。首先,在一组数据集中,如果某个样本的DNA甲基化谱中有10%以上的位点的检测值缺失,则去除这个样本;对于缺失率低于10%的甲基化谱,其缺失位点的DNA甲基化水平采用k近邻方法(k=1)补齐。然后,根据Iancu等[21]介绍的剔除异常样本方法,对每组数据的异常样本进行剔除。该方法简介如下:(1)基于每个样本检测的所有CpG位点的甲基化水平,计算每两个样本间的皮尔森相关系数;(2)计算每个样本与其他样本间的相关系数的均值即平均相关系数;(3)将所有样本的平均相关系数的均值与每个样本的平均相关系数进行比较,如果一个样本与其他样本的平均相关系数处于所有样本平均相关系数的两倍标准差之外,则作为异常样本剔除。对每组数据集重复上述异常样本剔除过程3次,以确保所有的异常样本都被去除。每组数据集中去除的样本数目见表1。
1.3 判定与老化相关的甲基化位点(arCpGs)
本文采用Spearman秩相关的方法计算每个CpG位点的甲基化水平改变和年龄的相关性[22],并利用BH(Benjamin and Hochberg)方法控制错误发现率(FDR)[23]。当一个位点的值经FDR校正后小于0.05时,该位点被定义为老化相关的甲基化位点(arCpGs)。由于统计效能不足,在一组数据中通常只能够找到部分的与老化相关的CpG位点。为了获得一个较全的arCpGs列表,本文将在Set1、Set2和Set3中分别找到的arCpGs合并在一起,然后再按如下规则剔除不可靠的arCpGs:(1)如果在一组数据集中识别的一个arCpG位点在其他两组数据集中的Spearman秩相关值都大于0.05,则剔除该位点;(2)如果一个位点在不同数据集中与年龄的正负相关性不同,亦剔除该位点。
在采用BH等方法控制多次检验的总体错误发现率时,通常会导致检验效能降低而难以在小样本数据中识别出显著差异的结果[24]。另一方面,仅以未经过校正的Spearman秩相关的值<0.05判断位点与年龄相关的显著性,又可能会出现过高的假阳性率。由于Set5和Set6分别只包含21和20个样本,本文利用“delete-1-jackknife[25]”方法识别这两组数据集中的arCpGs。采用“delete-1-jackknife”方法筛选在样本构成小幅度改变的条件下显著性水平(Spearman秩相关值<0.05)相对稳定的arCpGs,虽然无法校正多次检验的总体FDR,但是可减少由于某个样本的异常而导致的假阳性位点。该方法简介如下:每次从数据集中去掉一个样本,在剩下的样本中利用Spearman秩相关的方法按<0.05显著性水平寻找arCpGs;当数据集中的每个样本都被去除过一遍之后,即重复了(为该数据集的总样本数)次实验后,如果一个位点在所有次实验中都被判定为arCpGs,则该位点定义为该数据集的arCpGs。
1.4 在不同数据集中识别的arCpGs的重复性和一致性
假设识别自不同数据集的两个arCpGs列表分别包含L和L个CpG位点,且交叠了个位点,那么在这两个列表中观察到至少个位点交叠的随机概率可以用累计超几何分布来估计,计算公式如下:

其中代表背景位点数目。当<0.05时,则判定这两个arCpGs列表具有非随机的重叠。
如果一个交叠位点的DNA甲基化水平在这两组数据集中与年龄的正负相关性相同,则定义该位点为在两组数据集中与老化相关的一致位点。假设两个arCpGs列表交叠的个位点中有个一致位点,那么这两个列表交叠位点的一致性为。一致性的随机概率利用公式(3)计算。

其中0表示一个CpG位点在两组数据集中正、负相关性相同的随机概率,即0.5。
1.5 估计外周血中髓性细胞和淋巴性细胞的比例
利用Houseman等[26]开发的去卷积算法,估计各个外周全血样本中髓性细胞和淋巴性细胞的比例。Houseman等通过单因素方差分析在Set4中筛选出在各白细胞亚型间DNA甲基化水平方差变异程度最大的500个位点,并证实采用这500个特征位点,通过去卷积算法对DNA甲基化谱进行分解可获得较准确的结果。因此,本文利用这500个位点作为外周白细胞亚型的特征位点进行去卷积分析。该算法简单用公式(4)表示:
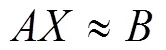
其中,代表特征位点在外周白细胞亚型中的DNA甲基化水平矩阵,表示每个外周血样本中各种白细胞亚型的比例,代表外周全血中的DNA甲基化谱数据矩阵。去卷积的目标是求得上述方程的解,即求得从而得到外周血各类型白细胞在每个样本中的比例[26]。然后,将来自髓性与淋巴性前体细胞的各种白细胞的比例分别相加,作为髓性与淋巴性细胞的比例。
1.6 判定髓性和淋巴性细胞间的差异甲基化位点
本文将Set4中的各类细胞按照髓性和淋巴性前体来源分为两组,再利用秩和检验判断位点在这两组细胞间的DNA甲基化水平发生差别的概率,并采用BH(Benjamin and hochberg)方法控制FDR。当一个位点的值经FDR校正后小于0.05时,判定该位点在两组细胞间显著差异。
1.7 老化相关甲基化位点的功能富集分析
利用Gene Ontology (GO)[27]的生物学过程(Biological process)功能注释体系对老化相关基因进行功能富集分析。首先,利用GEO提供的DNA甲基化芯片平台Illumina Infinium Human Methylation27 BeadChip的注释文件GPL8490-65.txt,将DNA甲基化位点注释到基因上。本文所分析的26 486个CpG位点共注释到13 890个基因(称为背景基因),并将其中与老化相关的位点注释到的基因称为老化相关的基因。
如果在个背景基因中,有L个基因与老化相关,其中个被注释到包含L个基因的GO功能结点中,那么随机情况下在这个GO功能结点中至少观察到个与老化相关的基因的概率可以通过累积超几何分布模型计算(公式2)。判定GO功能结点显著的标准是该节点的值经FDR校正后小于0.05。
2 结果与分析
2.1 外周全血中与老化相关的DNA甲基化改变的可重复性
利用Spearman秩相关分析,控制FDR<0.05,本文从数据集Set1、Set2和Set3中分别找到1239、1481、128个arCpGs,再将这3个arCpGs列表进行两两配对比较,结果如表2所示。任意两个列表的arCpGs都显著交叠(超几何检验,<1.65×10-11),且交叠部分的CpG位点的DNA甲基化水平与年龄的正、负相关性在两个列表中完全一致(二项分布检验,<2.2×10-16),提示在不同数据集中找到的与年龄相关的DNA甲基化位点存在显著的可重复性[28]。因此,本文利用1.3中介绍的方法,将3组数据集各自找到的arCpGs整合在一起,形成了一个包含1461个CpG位点的外周全血arCpGs列表,其中860个CpG位点与年龄正相关,601个CpG位点与年龄负相关。

表2 在不同数据集中识别的arCpGs比较
2.2 外周血细胞构成比例变化对外周全血中的arCpGs的影响
利用去卷积算法估计数据集Set1-3中的各个外周血样本中髓性和淋巴性细胞比例后,本文采用Spearman秩相关检验分析了髓性和淋巴性细胞的比例与年龄的相关性。结果显示,在这3组数据集中,髓性细胞与淋巴性细胞的比例变化与年龄分别呈现显著的正、负相关关系(Spearman秩相关检验,<0.05,表3),即外周全血中髓性与淋巴性细胞的比例分别随着年龄增大而增加、减少。在此前提下,若一个CpG位点在髓性细胞中的甲基化水平显著高于或低于在淋巴性细胞中的甲基化水平,即使该CpG位点在各类细胞中的DNA甲基化水平与年龄不相关,也可在外周全血中观察到该位点的甲基化水平与年龄显著地正相关或负相关[16]。在控制FDR<0.05的水平下,本文首先从Set4中识别了7184个DNA甲基化水平在髓性和淋巴性细胞间显著差异的CpG位点,记为mlCpGs。将外周全血中的arCpGs和mlCpGs比较,发现在外周全血中识别的1461个arCpGs中有1043(71.4%)个arCpGs与mlCpGs重叠,其中有798个位点(76.5%)的DNA甲基化水平在外周全血中与年龄的正、负相关性和其在髓性细胞中相对于淋巴性细胞的差异高、低甲基化不对应。该结果提示,大部分在外周全血中观察到的arCpGs并非由髓性与淋巴性细胞构成比例变化决定,而是反映白细胞内部的DNA甲基化改变。
另一方面,如图1所示,当CpG位点的DNA甲基化水平在髓性和淋巴性细胞间的差异程度大于0.3与0.35时,mlCpGs和外周全血中的arCpGs分别交叠了44与27个位点,它们与年龄的正、负相关性和其在髓性相对于淋巴性细胞间的差异甲基化方向的一致性分别提高到81.8%与92.6%,具有显著的非随机性(二项分布检验显著水平分别为<1.27×10–5,<2.82×10–6)。该结果提示,在外周全血中观察到的部分arCpGs主要受髓性和淋巴性细胞比例变化的影响,反映这两类细胞间的差异DNA甲基化信号。
2.3 外周全血中的arCpGs与CD4+T细胞和CD14+单核细胞中的arCpGs比较
外周全血中大部分的arCpGs可能反映白细胞亚型内部的DNA甲基化改变。因此,本文进一步分析外周全血中的arCpGs与在白细胞亚型中识别的arCpGs的关系。受数据限制,只能以淋巴性前体细胞来源的CD4+T细胞和髓性前体细胞来源的CD14+单核细胞为例进行初步分析。由于这两组数据集检测的样本数量过少,在FDR<0.05时,本文在检测CD4+T细胞的数据集Set5和检测CD14+单核细胞的数据集Set6中未找到arCpGs。因此,利用类似“delete-1-jackknife”的方法[25],分别在Set5和Set6数据集中找到了901个CD4+T细胞arCpGs和831个CD14+单核细胞arCpGs,这两个arCpGs列表只随机交叠了24个位点(超几何检验,=0.822),其中有21.7%的位点与年龄的正、负相关性在这两种细胞中不一致,提示这两种细胞的arCpGs可能不同。

表3 外周血中髓性细胞和淋巴细胞比例与年龄的相关性
注:*代表Spearman相关性的显著性水平<0.05。
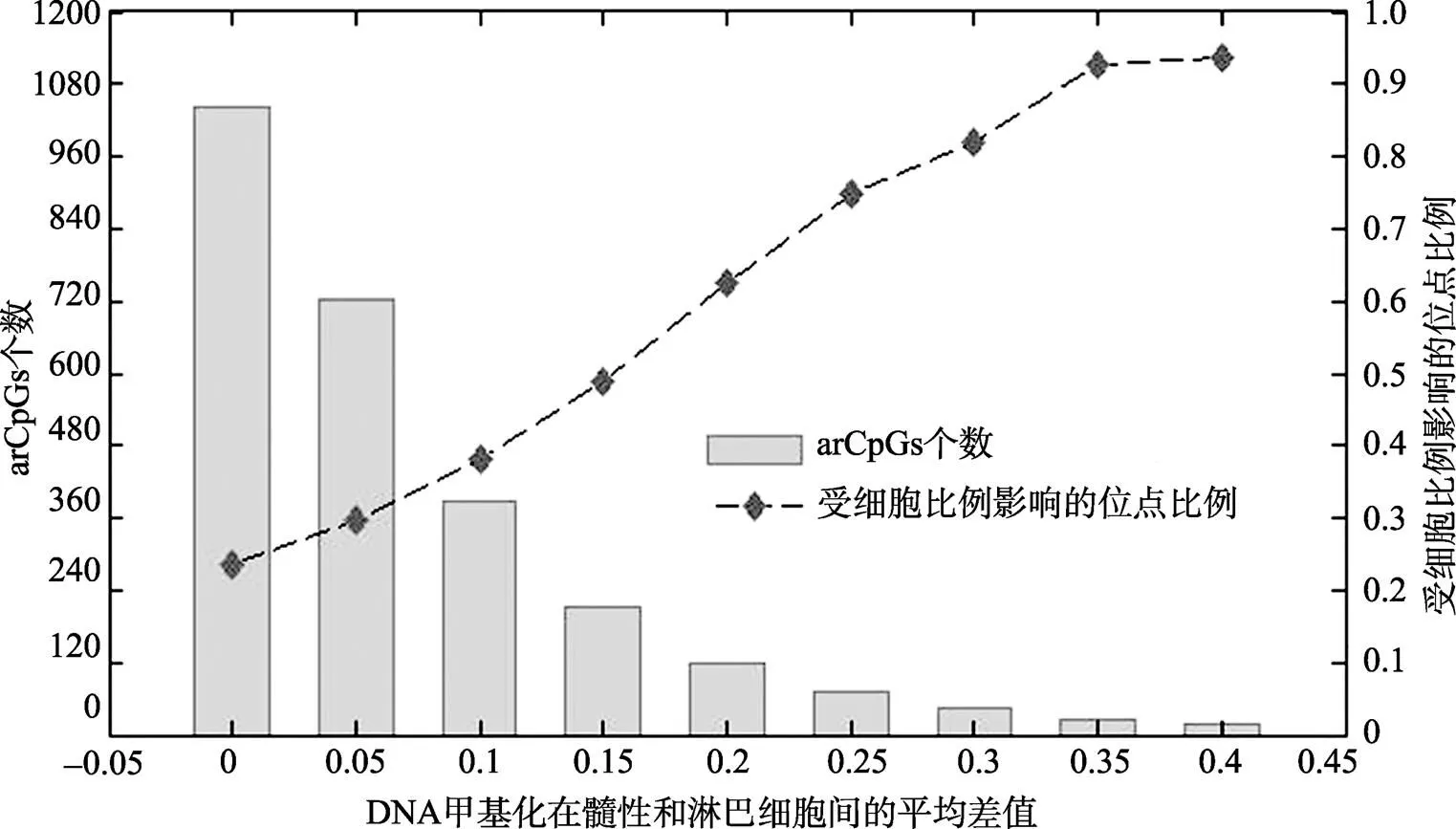
图1 细胞比例构成变化对外周全血中的arCpGs的影响
图中横坐标表示CpG位点的DNA甲基化水平在髓性和淋巴性细胞间的平均差值;左纵坐标表示在横坐标对应的平均差值下,识别的与年龄相关的CpG位点个数;右纵坐标表示在横坐标对应的平均差值下,外周全血arCpGs与年龄正、负相关性与其在髓性和淋巴性细胞间差异方向一致的比例。
分别将CD4+T细胞和CD14+单核细胞的arCpGs与外周全血arCpGs比较。结果显示,CD4+T细胞arCpGs与外周全血arCpGs非随机交叠了217个位点(超几何检验,=6.14×10-12),其中有99.1%的位点的DNA甲基化水平在CD4+T细胞和外周全血中与年龄的正、负相关性相同,呈现非随机高的一致性(二项分布检验,<2.2×10-16),说明CD4+T细胞中的一部分arCpGs能够在外周全血中反映。然而,CD14+单核细胞arCpGs只与外周全血arCpGs随机交叠了51个位点(超几何检验,=0.232),其中有48个位点的DNA甲基化水平与年龄正、负相关性在CD14+单核细胞和外周全血中一致(二项分布检验,=9.83×10-12)。值得注意的是,在这48个位点中,有46个位点在CD14+单核细胞和CD4+T细胞中与年龄的正、负相关性一致,提示这些位点可能是细胞间共同的与年龄相关的甲基化改变[19]。
在FDR<0.05的条件下,分别对在这两种细胞中识别的arCpGs进行GO功能富集。结果显示,CD4+T细胞arCpGs注释到的670个基因显著富集于20个GO功能结点,主要涉及发育过程、多细胞过程和细胞分化等相关的功能类(表4)。本文还发现,这20个GO功能结点也都显著富集了外周全血中的arCpGs (FDR<0.05),提示在外周全血中能够发现CD4+T细胞中的arCpGs涉及的大部分功能。另一方面,在FDR<0.05的条件下,CD14+单核细胞arCpGs注释到的610个基因没有富集到任何GO功能结点,提示在CD14+单核细胞中识别的arCpGs缺乏功能集聚特征。以上结果进一步提示这两种细胞的arCpGs可能存在差异。
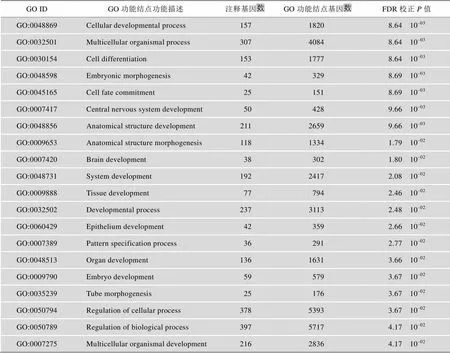
表4 CD4+T细胞中arCpGs所注释基因富集到的GO功能结点
3 讨 论
本文结果显示在外周全血中存在着稳定的与老化相关的DNA甲基化标记。外周全血中髓性细胞和淋巴性细胞的比例分别与年龄显著地正、负相关,这可能反映了随着年龄的增长,造血干细胞更趋向于分化为髓性细胞[7]。但是,由于细胞比例与年龄的相关系数的绝对值较小,仅为0.16~0.22(表3),在两类细胞间DNA甲基化水平差异程度较小的arCpGs主要反映白细胞内部的DNA甲基化改变。但是,仍然有一部分在髓性细胞和淋巴性细胞间DNA甲基化水平差异较大的位点倾向于在外周全血中被识别为arCpGs(即这部分arCpGs由细胞构成比例改变决定)。虽然Xu等[9]也分析过外周血白细胞比例变化对外周全血中的arCpGs的影响,但是由于他们仅评估了50个位点,未能发现本文的一个重要的结论,即在这两类细胞间DNA甲基化水平差异较大的位点倾向于在外周全血中被识别为arCpGs。由于目前尚缺乏伴有年龄信息的外周血各种白细胞亚型的DNA甲基化数据,本文只分析了CD4+T细胞和CD14+单核细胞中的arCpGs。值得注意的是,虽然CD4+T细胞只占外周白细胞总量的13%左右,但是许多CD4+T细胞的arCpGs却能在外周全血中被观察到。由于淋巴性细胞亚型间DNA甲基化模式相似,而淋巴性和髓性细胞间甲基化模式差异较大[15,16],对此现象的一种解释是其他淋巴性细胞上可能发生了与CD4+T细胞相似的改变。和CD4+T细胞不同,CD14+单核细胞的arCpGs与外周全血中的arCpGs没有显著交叠。但是,在这些交叠的arCpGs中,有90.1%的位点在CD14+单核细胞和全血中与老化的相关性一致,并且在CD4+T细胞中与老化的相关性也一致,提示这些位点可能反映了细胞间共同的与年龄相关的变化。Rakyan等[19]仅分析了CD4+T细胞与CD14+单核细胞共同识别的部分arCpGs,也发现了两者交叠且一致的arCpGs在外周全血中表现为与老化相关。本文对这两种细胞的arCpGs的比较分析进一步提示了这两种细胞可能存在各自特异的arCpGs,同时本文也评估了它们对在外周全血中观察到的arCpGs的影响。对在CD14+单核细胞中识别的大部分arCpGs未能在全血中反映的现象存在多种可能的解释:(1)检测该细胞的样本太少,导致识别其arCpGs的统计效能不足或假阳性结果过多;(2)该细胞在外周血白细胞中所占比例较小(6%左右),其大部分arCpGs未在其他(髓性)细胞出现,因此这些arCpGs信号可能被其他细胞上的arCpGs信号所掩盖;(3)由于相对于淋巴性细胞,髓性细胞在外周血中的存活时间很短[29],因此髓性前体细胞分化时出现髓性细胞特异的年龄相关的DNA甲基化改变的机会较小。然而,这些推测还需要更多的实验数据来进一步验证。
老化相关的分子改变与癌症等老化相关疾病的发病风险相关[1]。本文的分析结果显示,外周全血中与老化相关的DNA甲基化改变主要反映白细胞内部及细胞比例构成的改变。但是,癌症外周血中的异常DNA甲基化信号主要由疾病状态下外周全血中髓性和淋巴性细胞的比例变化决定[16]。因此,在分析外周全血中的arCpGs和疾病的关系时,应该首先排除外周血细胞构成比例变化对疾病相关标记的影响。
[1] Ahuja N, Issa JP. Aging, methylation and cancer, 2000, 15(3): 835–842.
[2] López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging, 2013, 153(6): 1194–1217.
[3] Vanyushin BF, Nemirovsky LE, Klimenko VV, Vasiliev VK, Belozersky AN. The 5-methylcytosine in DNA of rats. Tissue and age specificity and the changes induced by hydrocortisone and other agents, 1973, 19(3): 138–152.
[4] Vanyushin BF, Tkacheva SG, Belozersky AN. Rare bases in animal DNA, 1970, 225(5236): 948–949.
[5] Wilson VL, Smith RA, Ma S, Cutler RG. Genomic 5-methyldeoxycytidine decreases with age, 1987, 262(21): 9948–9951.
[6] Heyn H, Li N, Ferreira HJ, Moran S, Pisano DG, Gomez A, Diez J, Sanchez-Mut JV, Setien F, Carmona FJ, Puca AA, Sayols S, Pujana MA, Serra-Musach J, Iglesias-Platas I, Formiga F, Fernandez AF, Fraga MF, Heath SC, Valencia A, Gut IG, Wang J, Esteller M. Distinct DNA methylomes of newborns and centenarians, 2012, 109(26): 10522–10527.
[7] Bocker MT, Hellwig I, Breiling A, Eckstein V, Ho AD, Lyko F. Genome-wide promoter DNA methylation dynamics of human hematopoietic progenitor cells during differentiation and aging, 2011, 117(19): 182–189.
[8] Horvath S, Zhang YF, Langfelder P, Kahn RS, Boks MPM, Van Eijk K, Van Den Berg LH, Ophoff RA. Aging effects on DNA methylation modules in human brain and blood tissue, 2012, 13(10): R97.
[9] Xu ZL, Taylor JA. Genome-wide age-related DNA methylation changes in blood and other tissues relate to histone modification, expression and cancer, 2014, 35(2): 356–364.
[10] Castelo-Branco C, Soveral I. The immune system and aging: a review, 2014, 30(1): 16–22.
[11] Djukic M, Nau R, Sieber C. The ageing immune system, 2014, 139(40): 1987–1990.
[12] Panda A, Arjona A, Sapey E, Bai F, Fikrig E, Montgomery RR, Lord JM, Shaw AC. Human innate immunosenescence: causes and consequences for immunity in old age, 2009, 30(7): 325–333.
[13] Golbus J, Palella TD, Richardson BC. Quantitative changes in T cell DNA methylation occur during differentiation and ageing, 1990, 20(8): 1869–1872.
[14] Vasson MP, Farges MC, Goncalves-Mendes N, Talvas J, Ribalta J, Winklhofer-Roob B, Rock E, Rossary A. Does aging affect the immune status? A comparative analysis in 300 healthy volunteers from France, Austria and Spain, 2013, 10(1): 38.
[15] Reinius LE, Acevedo N, Joerink M, Pershagen G, Dahlen SE, Greco D, Söderhäll C, Scheynius A, Kere J. Differential DNA methylation in purified human blood cells: implications for cell lineage and studies on disease susceptibility, 2012, 7(7): e41361.
[16] Li H, Zheng T, Chen B, Hong G, Zhang W, Shi T, Li S, Ao L, Wang C, Guo Z. Similar blood-borne DNA methylation alterations in cancer and inflammatory diseases determined by subpopulation shifts in peripheral leukocytes, 2014, 111(3): 525–531.
[17] Barrett T, Troup DB, Wilhite SE, Ledoux P, Rudnev D, Evangelista C, Kim IF, Soboleva A, Tomashevsky M, Marshall KA, Phillippy KH, Sherman PM, Muertter RN, Edgar R. NCBI GEO: archive for high-throughput functional genomic data, 2009, 37(S1): D885–D890.
[18] Accomando WP, Wiencke JK, Houseman EA, Butler RA, Zheng S, Nelson HH, Kelsey KT. Decreased NK cells in patients with head and neck cancer determined in archival DNA, 2012, 18(22): 6147–6154.
[19] Rakyan VK, Down TA, Maslau S, Andrew T, Yang TP, Beyan H, Whittaker P, Mccann OT, Finer S, Valdes AM, Leslie RD, Deloukas P, Spector TD. Human aging-associated DNA hypermethylation occurs preferentially at bivalent chromatin domains, 2010, 20(4): 434– 439.
[20] Brock MV, Hooker CM, Ota-Machida E, Han Y, Guo MZ, Ames S, Glockner S, Piantadosi S, Gabrielson E, Pridham G, Pelosky K, Belinsky SA, Yang SC, Baylin SB, Herman JG. DNA methylation markers and early recurrence in stage I lung cancer, 2008, 358(11): 1118– 1128.
[21] Iancu OD, Darakjian P, Walter NAR, Malmanger B, Oberbeck D, Belknap J, McWeeney S, Hitzemann R. Genetic diversity and striatal gene networks: focus on the heterogeneous stock-collaborative cross (HS-CC) mouse, 2010, 11: 585.
[22] Alisch RS, Barwick BG, Chopra P, Myrick LK, Satten GA, Conneely KN, Warren ST. Age-associated DNA methylation in pediatric populations, 2012, 22(4): 623–632.
[23] Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing1995, 57(1): 289–300.
[24] Zhang M, Yao C, Guo Z, Zou JF, Zhang L, Xiao H, Wang D, Yang D, Gong X, Zhu J, Li YH, Li X. Apparently low reproducibility of true differential expression discoveries in microarray studies, 2008, 24(18): 2057– 2063.
[25] [Qiu X, Xiao YH, Gordon A, Yakovlev A. Assessing stability of gene selection in microarray data analysis, 2006, 7: 50.
[26] Houseman EA, Accomando WP, Koestler DC, Christensen BC, Marsit CJ, Nelson HH, Wiencke JK, Kelsey KT. DNA methylation arrays as surrogate measures of cell mixture distribution, 2012, 13: 86.
[27] Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium, 2000, 25(1): 25–29.
[28] Zhang M, Zhang L, Zou JF, Yao C, Xiao H, Liu Q, Wang J, Wang D, Wang CG, Guo Z. Evaluating reproducibility of differential expression discoveries in microarray studies by considering correlated molecular changes, 2009, 25(13): 1662–1668.
[29] Yang J, Zhang LX, Yu CJ, Yang XF, Wang H. Monocyte and macrophage differentiation: circulation inflammatory monocyte as biomarker for inflammatory diseases, 2014, 2(1): 1.
(责任编委: 方向东)
Age-related DNA methylation changes in peripheral whole blood
Hongdong Li1, Guini Hong1, Zheng Guo1,2
Aging is associated with many complex diseases such as cancer and neurodegenerative diseases. Recently, many age-related DNA methylation biomarkers in peripheral whole blood have been identified. These biomarkers may reflect DNA methylation changes derived from changes in the number of a specific leukocyte cell type during aging. To clarify the source of these age-related DNA methylation changes, we analysed DNA methylation profile of peripheral whole blood from three independent cohorts of healthy subjects and identified age-related DNA methylation CpG sites (arCpGs) using the Spearman’s rank test with high reproducibility (Hypergeometric test,=1.65×10-11). Using a deconvolution algorithm, we found that the proportion of myeloid lineage cells was increased while that of lymphoid lineage cells was decreased in the peripheral whole blood with age (Spearman’s rank correlation test,<0.05,≤0.22). The CpG sites, whose methylation levels were significantly different in myeloid cells and lymphoid cells, were preferentially recognized as arCpGs in peripheral whole blood.Moreover, the arCpGs in CD4+ T cells significantly overlapped with that in peripheral whole blood (Hypergeometric test,=6.14×10-12) and 99.1% of the overlapping arCpGs had consistent positive or negative correlations with age. Though the arCpGs in CD14+ monocytes did not significantly overlap with that in peripheral whole blood (Hypergeometric test,=0.232), 90.1% of 51 overlapping arCpGs were correlated with age in CD14+ monocytes, peripheral whole blood, and CD4+ T cells consistently. In summary, most of the methylation changes in arCpGs identified in peripheral whole blood come from common or specific DNA methylation changes in leukocyte subtypes, while part of them reflect alterations in the number of specific cell types of leukocytes.
peripheral whole blood; aging; DNA methylation; CD4+ T cells; CD14+ monocytes
2014-11-12;
2014-11-20
国家自然科学基金项目(编号:81372213,81201822)资助
李红东,博士研究生,研究方向:生物信息学。Tel: 028-83207187;E-mail: biomantis_lhd@163.com
郭政,博士,教授,研究方向:生物信息学。E-mail: guoz@ems.hrbmu.edu.cn
10.16288/j.yczz.14-394
网络出版时间: 2014-10-15 8:06:17
URL: http://www.cnki.net/kcms/detail/11.1913.R.20141015.0806.002.html
猜你喜欢
中学生数理化·中考版(2021年9期)2021-11-20
世界最新医学信息文摘(2021年12期)2021-06-09
甘肃教育(2020年4期)2020-09-11
家庭医药·快乐养生(2019年2期)2019-03-04
作文周刊·小学四年级版(2016年1期)2016-04-15
医学研究杂志(2015年12期)2015-06-10
中国医科大学学报(2015年10期)2015-03-01
癌变·畸变·突变(2015年3期)2015-02-27
中国信息化周报(2015年4期)2015-02-09
中国海洋大学学报(自然科学版)(2014年12期)2014-02-28
