
基于血凝素裂解位点的DNA条形码在A型流感病毒分型中的应用
2015-12-16 07:48孟慧芝岳志芹王文斌
中国动物检疫 2015年7期
孟慧芝,孙 涛,岳志芹,王文斌
(1.山东出入境检验检疫局,山东青岛 266000;2.山西农业大学,山西太谷 030801)
基于血凝素裂解位点的DNA条形码在A型流感病毒分型中的应用
孟慧芝1,2,孙 涛1,岳志芹1,王文斌2
(1.山东出入境检验检疫局,山东青岛 266000;2.山西农业大学,山西太谷 030801)
为了建立一种快速鉴别AIV的HA亚型的RT-PCR方法,本研究探讨了流感病毒血凝素裂解位点基因作为DNA条形码对A型流感病毒进行亚型鉴定的可行性。通过生物信息学方法对16种A型流感病毒的血凝素基因全长进行比对,在HA裂解位点的两端高度保守区设计一对DNA条形码引物,RT-PCR扩增获得约170bp的核苷酸片段。利用MEGA5.0软件构建N-J系统树并计算亚型间及亚型内平均遗传距离。结果显示,不同A型流感病毒裂解位点氨基酸具有单系性,每一亚型可分别形成各自独立的分支。不同亚型间平均遗传距离为0.277,相同亚型内平均遗传距离0.032。研究表明,利用DNA 条形码技术可在A型流感病毒分型方面进行有效的分子分类鉴定。
A型流感病毒;血凝素(HA);DNA条形码;亚型鉴定
根据表面糖蛋白血凝素(hemagglutinin,HA)和神经氨酸酶(neuraminidase,NA)的抗原性差异,A型流感病毒可分为18种HA亚型和11种NA亚型[1-2]。Alexander[3]在对16%野生水禽原始泄殖腔样品检测时发现,A型流感病毒存在种群的准种,存在两到五个不同血凝素亚型混合感染,流感病毒变异快、宿主范围广,每年暴发的流感差异也表明流感在不同宿主间交叉传播愈加频繁[4],给病毒的全面准确检测带来了极大的挑战。
DNA 条形码技术是近年来发展最迅速的学科之一[5],它通过使用一段标准 DNA片段,可对物种或型别进行快速、准确地鉴定。其工作的核心是
寻找适合的条形码序列,一方面足够保守能够利用通用引物进行大范围扩增,另一方面足够变异能够区分密切相关的物种或亚型[6]。
Lee[7]等人曾用coI基因鉴别出了含禽流感病毒的26种野生鸟类,为研究候鸟流感流行病学的规律和宿主种群生态学提供依据。但对病毒亚型本身的条码技术开发尚未见相应报道,因此本研究拟尝试开发更加广谱、便捷的通用流感病毒监测技术用以监测流感病毒。
1 材料和方法
1.1 毒株及载体
猪流感H1N1,马流感H3N8,禽流感H2N9、H4N1、H5N1、H7N2、H9N2、H13N6、H16N3病毒核酸样本由本实验室保存;H6N5、H8N4、H10N8、H11N6、H12N5、H14N5、H15N8病毒核酸样本由吉林省出入境检验检疫局惠赠(具体毒株信息见表1);16份毒株的HA亚型采用常规HI抗体血清反应进行确认;DH5α大肠杆菌感受态细胞及pMD-18T克隆载体为Takara公司产品。
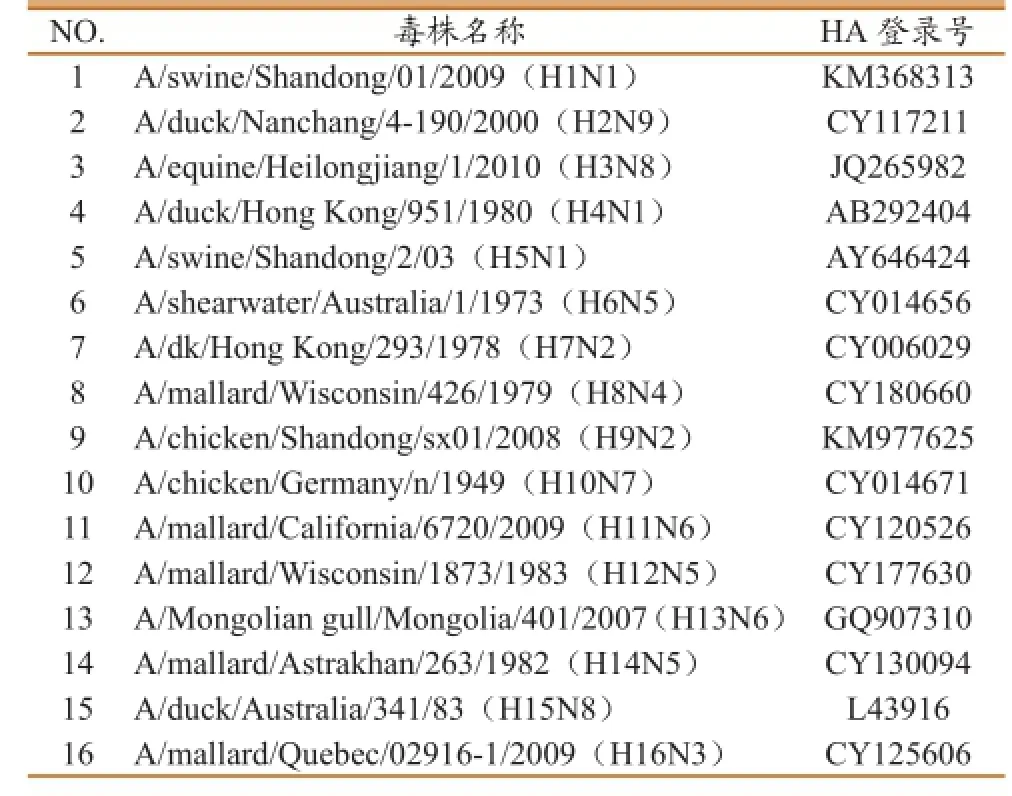
表1 本研究所用的A型流感病毒株序列信息
1.2 分子生物学试剂
Rneasy Mini Kit为QIAGen公 司 产 品;1 st strand cDNA Synthesis Kit cDNA反转录试剂盒、EX-Taq聚合酶、Solution Ⅰ连接酶、Agarose Gel DNA Extration Kit试剂盒、Plasmid Purifi cation Kit试剂盒均为Takara公司产品。
1.3 基因组提取及反转录
按QIAGen RNA提取试剂盒操作步骤提取RNA,每份病毒RNA样品溶解于50μL的Rnasefree ddH2O,1 2000 r/min离心2 min,立即开始反转录,避免RNA降解。以U12 5'-agcaaaagcagg-3'为引物[8],参照Takara公司1 st strand cDNA Synthesis Kit cDNA试剂盒使用说明加入试剂,RT反应条件为:30℃10min,42℃40 min,70 ℃15min,-20 ℃保存。
1.4 引物设计与PCR扩增
在NCBI流感病毒库中,批量下载16种亚型的HA的全长序列,每个亚型50条(除H16),同一区域同一年代只选一个代表株,通过MEGA5.0软件的Align程序比对所有HA亚型全长基因,寻找流感病毒HA的标志序列,设计DNA条码引物并送invitrogen公司合成。上游引物:GGRRRATGCCCCARRTAYRT,下游引物:CGTA GTCCGAACATTGAGTTBYKATGNYKRWADCCR TACCA。因扩增产物G+C含量百分比不同,退火温度会有差异,故PCR反应采用touch-down程序:94 ℃2 min,{94 ℃ 30 s,53 ℃30 s每个循环递减1 ℃直到45 ℃;68 ℃ 45 s}8个循环。{94 ℃ 30 s,50 ℃ 30 s,68 ℃ 45 s}30个循环,68 ℃10 min。-20 ℃保存。
1.5 连接与转化
PCR产物的回收按Agarose Gel DNA Purifi cation Kit 说明书进行。将各自胶回收的PCR产物与pMD-18T载体1∶3比例16℃过夜连接后,取10μL加入200μL的感受态细胞悬液,42 ℃热激1.5 min,冰上迅速冷却,加入1 mL不含Amp LB液体培养基混匀,37 ℃振荡培养1 h,取50μL菌液涂布于含氨苄的LB培养基上,37 ℃过夜培养,挑取生长状况良好的单个菌落,再接种于含氨苄的LB液体培养基中,37 ℃过夜培养。
刘勰《文心雕龙》中,标“隐秀”一格,“是以文之英蕤,有秀有隐。隐也者,文外之重旨者也;秀也者,篇中之独拔者也。隐以复意为工,秀以卓绝为巧”⑱,故所谓“隐秀感”,有两层含义:其一,显与隐的纵深关系,“秀”为显,“隐”为“秀”的浑茫背景,笔者曾提出“生动在浑茫的背景里”,恰为此义;其二,奇与正的横向关系,“秀”为奇,“隐”为正,整体之“隐”,衬托独拔之“秀”。此原理中,“隐”接通无、浑茫的模糊,“隐”与“秀”平衡的分寸,皆是幽微玄妙,全凭感悟而得,因此独擅此道的中国古典艺术批评,深富“隐秀感”。
1.6 测序及比对
为了评估扩增产物的准确性,经鉴定为阳性菌落挑克隆培养菌液送宝生物测序,BLAST搜索比对,验证所扩增片段与已知HA各亚型的同源性和
准确性。
1.7 系统进化分析与遗传距离计算
将测定的序列与GenBank中参考序列在MEGA5.0的CLAST中比对,除去两条引物之外的序列,基于邻接法(neighbor-joining method)构建系统进化树,并且计算遗传距离。运算次数为1 000,核酸替换模型为采用国际提倡的K-2P,K-2P距离与P距离相比考虑了转换和颠换的替代率,是在变异很小时区分物种的最佳模型也是生物条形码联盟推荐使用的距离计算模型。
2 结果
2.1 RT-PCR结果
16种不同亚型的毒株RT-PCR均能扩增出164~176 bp的目的片段,经1%凝胶电泳,与预期大小相符(图1)。

图1 16株不同实验株HA裂解位点RT-PCR扩增结果
代表16个不同亚型的16个流感病毒测试样品,经BLAST搜索结果证实基于HA裂解位点的DNA条形码所识别亚型与全长测序一致。
2.3 16株毒株的裂解位点氨基酸分析结果(图2)
裂解位点的氨基酸翻译分析显示:H5N1有明显的AGAAAAAAGCGA插入,翻译蛋白为RKKR。裂解位点内发现有连续多个碱性氨基酸,为高致病性AIV的分子特征。原病毒经哈尔滨兽医研究所国家禽流感参考实验室检测,病毒静脉接种致病指数(IVPI)鉴定结果为2.6,符合OIE规定的静脉致病的1.2以上的强毒特征,其他测试毒株均为低致病性毒株。

图2 16株实验株HA裂解位点氨基酸位点分析结果
2.4 HA0分子进化树分析结果
选择与每个HA结合的不同NA亚型代表株构建N-J进化树,本实验所用毒株前面均加三角号,由图3、图4可知,所有A型流感病毒的HA裂解位点处核苷酸序列均有一个共同的祖先,H1、H2、H3、H4、H5、H6、H8、H9、H10、H11、H12、H13、H14、H16、H17、H18均能形成各自独立的分支。但对于H7和H15亚型,由于亲缘关系较近,种间差异较小,种内和种间的遗传变异存在重叠。而对于氨基酸序列而言,每个DNA条形码扩增所得的氨基酸序列,均能在不同HA亚型的个体间形成单系,并能帮助准确识别亚型多样性。
2.5 遗传距离计算
由于基于所选区域核酸序列选择K-2P遗传距离仍然无法区分H7和H15,两者之间亲密度很高,所以选用蛋白序列来计算遗传距离,采用MEGA5.0 中的Distance 模块,Variance Estimation Method设置为Bootstrap method,1 000次,氨基酸替换模型为P-distance。所得裂解位点的蛋白遗传距离,H1-H16亚型内的遗传距离最大值分别为:0.121、0.085、0.155、0.068、0.136、0.119、0.121、0.068、0.186、0.083、0.068、0.102、0.085、0.034、0.033、0.051,H1、H5、H7、H9遗传距离变化范围较大,可能是这些亚型宿主范围广,遗传变异复杂,多个分支并存所致[9-10]。而H4、H14、H15仅在野生鸟类分离到变化幅度较小。H1-H16亚型内的平均遗传距离分别为:0.038、0.026、0.046、0.015、0.054、0.038、0.049、0.024、0.060、0.041、0.020、0.033、0.025、0.011、0.014、0.020。亚型内遗传距离平均
值为0.032,16种不同亚型间平均遗传距离为0.277。
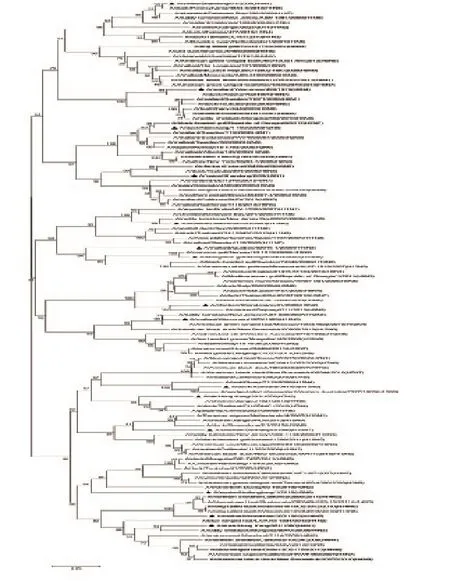
图3 16种不同A型流感亚型裂解位点核酸序列比对图
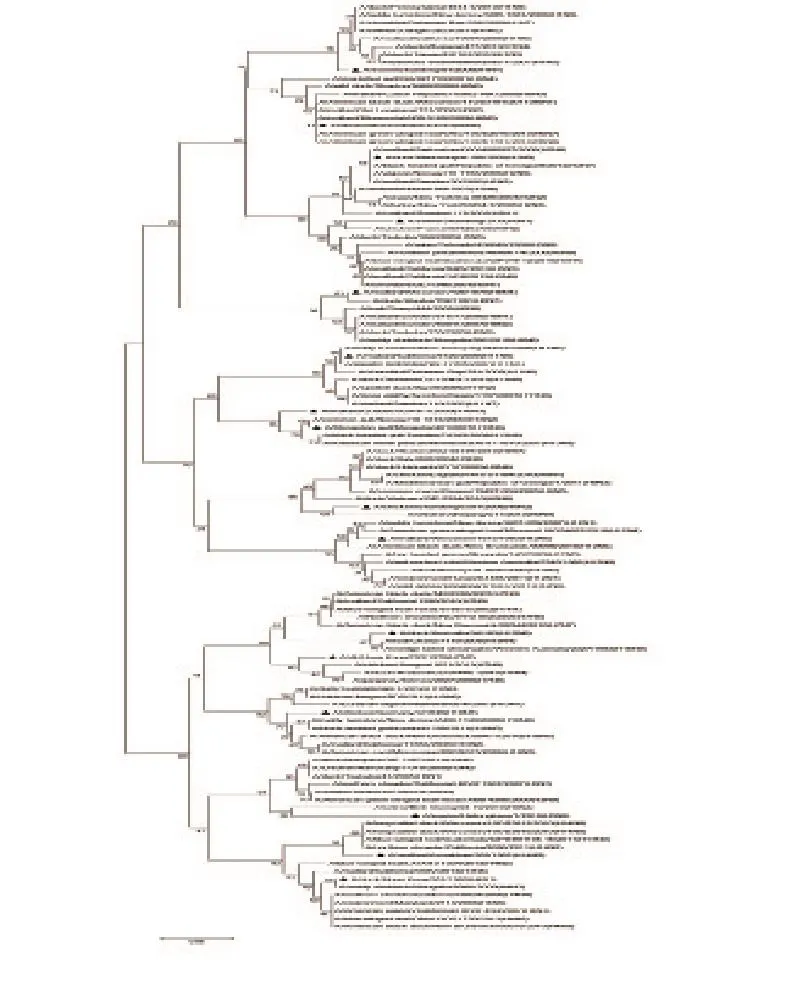
图4 16种不同A型流感亚型裂解位点氨基酸序列比对图
3 讨论
本研究通过对不同宿主、地域的16种A型流感病毒的HA0裂解位点序列进行分析,发现裂解位点处氨基酸序列可作为区分各亚型的DNA条码,比传统方法更具敏感性和特异性。Hebet[5]曾提出利用条形码序列有效鉴别的关键点是种间的遗传距离必须大于种内,且距离差异大约为10倍。本研究中的型间平均遗传距离为0.277,高于型内平均遗传距离0.032。分析系统树显示,各亚型均能独立形成单系。
目前,实验室广泛推广的流感检测方法是基于M基因或NP基因进行的TaqMan实时荧光PCR检测方法[11-12],但常规诊断存在对H5、H7以及其他亚型的RT-PCR检测逃避[13]。实时荧光定量RTPCR实验对HA亚型特异性鉴定不完整现象,不能完全鉴定16种亚型。而通过PCR和进一步的测序程序是全面确定样本中HA亚型的有力证据。Hoffman等曾设计引物[8]采用全基因扩增进行流感鉴定,但需要大量优化,产物得率较低且杂带较多,不适于实际的常规监测或诊断[14]。基因芯片技术[15]则需专业设备和人员,难以推广;本文利用DNA条形码技术思路,采用传统RT-PCR检测方法,优选touch-down程序可以快速判定有无流感病毒,并能同时测定A型流感病毒的亚型及致病型,是一种更简单,快速的流感分型诊断程序。
HA0裂解位点的氨基酸序列是AIV致病力的分子基础,直接影响病毒致病力的高低[16-17]。通常情况下,高致病性禽流感病毒在HA0裂解位点处具有多个精氨酸和赖氨酸,并有明显的插入或替代[18],可被多种蛋白酶裂解[19]。从高致病性禽流感流行历史来看,随着病毒的不断扩散和对宿主适应性的增强,病毒的毒力可通过未知的机制发生改变而对禽类的致病力增强,高致病力毒株均由低致病性病毒的前体发生突变导致[20-21]。因此,本文利用DNA条形码技术对流感病毒各亚型的裂解位点进行分析,可为及时处理传染性材料的风险提供有效手段[22]。
[1] Tong S,Li Y,Rivailler P,et al. A distinct lineage of infl uenza A virus from bats[J]. Proc Natl Acad Sci U S A,2012,109(11):4269-4274.
[2] Tong S,Zhu X,Li Y,et al. New world bats harbor diverse infl uenza A viruses[J]. PLoS Pathog,2013,9(10):e1003657.
[3] Wang R,Soll L,Dugan V,et al. Examining the hemagglutinin subtype diversity among wild duck-origin influenza A viruses using ethanol-fixed cloacal swabs and a novel RT-PCR method[J]. Virology,2008,375(1):182-189.
[4] Alexander D J. A review of avian infl uenza in different bird species[J]. Vet Microbiol,2000,74(1/2):3-13.
[5] Hebert P D,Cywinska A,Ball S L,et al. Biological identifi cations through DNA barcodes[J]. Proc Biol Sci,2003,270(1512):313-321.
[6] Lahaye R,van der Bank M,Bogarin D,et al. DNA barcoding the fl oras of biodiversity hotspots[J]. Proc Natl Acad Sci U S A,2008,105(8):2818-2923.
[7] Lee D H,Lee H J,Lee Y J,et al. DNA barcoding techniques for avian influenza virus surveillance in migratory bird habitats[J]. J Wildl Dis,2010,46(2):649-654.
[8] Hoffmann E,Stech J,Guan Y,et al. Universal primer set for the full-length amplifi cation of all infl uenza A viruses[J]. Arch Virol,2001,146(12):2275-2289.
[9] Chen H,Smith G J,Li K S,et al. Establishment of multiple sublineages of H5N1 influenza virus in Asia:implications for pandemic control[J]. Proc Natl Acad Sci U S A,2006,103(8):2845-2850.
[10] Keawcharoen J,Oraveerakul K,Kuiken T,et al. Avian influenza H5N1 in tigers and leopards[J]. Emerg Infect Dis,2004,10(12):2189-2191.
[11] Hoffmann B,Depner K,Schirrmeier H,et al. A universal heterologous internal control system for duplex real-time RTPCR assays used in a detection system for pestiviruses[J]. J Virol Methods,2006,136(1/2):200-209.
[12] Kiss I,German P,Sami L,et al. Application of real-time RT-PCR utilising lux(light upon extension) fl uorogenic primer for the rapid detection of avian influenza viruses[J]. Acta Vet Hung,2006,54(4):525-533.
[13] Anonymous. Chapter 2.5.7 Equine influenza[M]//OIE Manual of diagnostic tests and vaccines for terrestrial animal. 2012,World Organization for Animal Health(Paris,France.).
[14] Li O T,Barr I,Leung C Y,et al. Reliable universal RTPCR assays for studying influenza polymerase subunit gene sequences from all 16 haemagglutinin subtypes[J]. J Virol
Methods,2007,142(1/2):218-222.
[15] 韩雪清,林祥梅,侯义宏,等. 禽流感病毒分型基因芯片的研制[J]. 微生物学报,2008,48(9):1241-1249.
[16] Weis W,Brown J H,Cusack S,et al. Structure of the influenza virus haemagglutinin complexed with its receptor,sialic acid[J]. Nature,1988,333(6172):426-431.
[17] Senne D A,Panigrahy B,Kawaoka Y,et al. Survey of the hemagglutinin(HA) cleavage site sequence of H5 and H7 avian infl uenza viruses:amino acid sequence at the HA cleavage site as a marker of pathogenicity potential[J]. Avian Dis,1996,40(2):425-437.
[18] Horimoto T,Kawaoka Y. Reverse genetics provides direct evidence for a correlation of hemagglutinin cleavability and virulence of an avian infl uenza A virus[J]. J Virol,1994,68(5):
3120-3128.
[19] Lee C W,Swayne D E,Linares J A,et al. H5N2 avian infl uenza outbreak in Texas in 2004:the fi rst highly pathogenic strain in the United States in 20 years[J]. J Virol,2005,79(17):11412-11421.
[20] Alexander D J. An overview of the epidemiology of avian infl uenza[J]. Vaccine,2007,25(30):5637-5644.
[21] Suarez D L,Senne D A,Banks J,et al. Recombination resulting in virulence shift in avian infl uenza outbreak,Chile[J]. Emerg Infect Dis,2004,10(4):693-699.
[22] Eames K T,Webb C,Thomas K,et al. Assessing the role of contact tracing in a suspected H7N2 infl uenza A outbreak in humans in Wales[J]. BMC Infect Dis,2010,10:141.
(责任编辑:胡藕祥)
Application of DNA Barcoding Based on Hemagglutinin Cleavage Site for Infl uenza A Virus Sub-typing
Meng Huizhi1,2,Sun Tao1,Yue Zhiqin1,Wang Wenbin2
(1.Shandong Entry-Exit Inspection and Quarantine Bureau,Qingdao,Shandong 266000;2.Shanxi Agricultural University,Taigu,Shanxi 030801)
The feasibility of the hemagglutinin cleavage sites used as DNA barcoding to subtype influenza A virus was explored in the study. According to the alignment of the hemagglutinin gene of 16 subtypes of infl uenza A virus by bioinformatics means,a pair of primers were designed on both terminals of highly conserved of hemagglutinin cleavage site.170bp products were obtained by RT-PCR. Then phylogenetic tree was constructed and the average intratype variation and inter-type divergence were calculated with MEGA5.0 software. The phylogenetic tree revealed that amino acid composition of cleavage site was of monophyly in each subtype. The average genetic distance of inter-type was 0.277,and intra-type genetic distance was 0.032. It was concluded that the hemagglutinin DNA barcoding can be used for subtyping infl uenza A virus effectively.
infl uenza A virus;hemagglutinin(HA);DNA barcoding technology;subtype
S852.65
A
1005-944X(2015)07-0055-07
国家科技支撑计划(2012BAK11B00)
孙 涛
猜你喜欢
广东农业科学(2022年11期)2023-01-13
少年文艺·开心阅读作文(2021年8期)2021-09-05
皮肤病与性病(2021年3期)2021-07-30
世界科学技术-中医药现代化(2021年12期)2021-04-19
中国动物检疫(2020年1期)2020-01-08
科学24小时(2019年5期)2019-06-11
小学科学(学生版)(2019年5期)2019-05-21
猪业科学(2018年6期)2018-01-23
学苑创造·B版(2017年10期)2017-12-21
