
常染色体显性遗传性耳聋*(1)
2016-04-13 05:11王秋菊,刘穹
听力学及言语疾病杂志 2016年2期
常染色体显性遗传性耳聋*(1)
王秋菊1刘穹1
网络出版时间:http://www.cnki.net/kcms/detail/42.1391.R.20160224.1502.004.html
网络出版地址:2016-2-2415:02
常染色体显性遗传性耳聋常常指的是非综合征型遗传性耳聋,是以耳聋为单一症状,不伴有全身其他器官系统的异常,且遗传基因位于常染色体上,并由显性基因所控制。最早的报道可追溯至17世纪,1814年,Adams等报道了一个四代受累的疑似耳硬化症的垂直传递的耳聋家系,其临床遗传学特征为:双亲之一是患者,子女中半数可发病;耳聋发生与性别无关,男女发病机会均等;可见连续几代的发病情况。在遗传性耳聋中,约22%为显性遗传,77%是隐性遗传,1%是X连锁、线粒体突变母系遗传或Y连锁遗传。
1常染色体显性遗传性耳聋致病基因的命名原则、定位方法和基因的发现
常染色体显性遗传性聋,英文表述为Autosomal Dominant Hereditary Hearing Loss,缩略词为ADHHL。谈及常染色体显性遗传性耳聋,要了解其命名原则,定位方法和致病基因,含以下三个方面:
1.1致病基因的座位英文表述为locus,复数为loci,是根据人类基因组命名委员会的规则,以DFNA来表示常染色体显性遗传性耳聋,DFN缩略词来自DeaFNess (耳聋)中的三个字母,A定义为显性遗传。第一个致病基因座位表述为DFNA1,第二个表述为DFNA2。基因座的命名常常是由遗传连锁分析的方法而获得的,由人类基因组命名委员会(Human Genome Organization nomenclatuer commitee HUGO Httpc/ /www.gen e.uc l.ac .uk /nomenclatuer/)来认可和发布,而基因座命名的申请是由研究者根据研究结果提交到命名委员会,通过命名委员会的审核后确定。命名的序号是根据发现时间的顺序以及获得命名的时间依次来排序的,到目前为止常染色体显性遗传性耳聋的基因座位是从DFNA1到DFNA67来排序的,如有新的发现还会依次增加序号。
1.2致病基因的位置英文表述为location,以染色体的序号,染色体的短臂(p)或长臂(q)以及定位的区段来表示。以DFNA1为例,1992年,Léon等利用限制性片段长度多态性(restriction fragment length polymorphisms RFLP)标记方法,将一个八代相传的哥斯达黎加家系的耳聋基因定位在5号染色体上长臂的3区1带上,由于这是第一个发现和定位的显性遗传性耳聋基因座位,因此申请命名为DFNA1基因座,其定位的位置表述为5q31。在这个定位区间有约7厘摩(centimogan, cM)的遗传距离,DFNA1连锁的区域有800 kb大小,因此定位之后的基因发现仍然为一个巨大的挑战,需要继续研究,故描述显性遗传性耳聋时的第三方面的内容就是致病基因。
1.3显性遗传性耳聋的致病基因英文表述为 pathogenic or causative gene,在人类基因组30亿个碱基对中发现一个耳聋致病基因是一个大海捞针的过程。DFNA基因发现方式常常是经过经典的连锁分析方法将致病基因定位在染色体上的一个区段,由此缩小了基因发现的范围,之后就是应用候选基因筛查发现其致病基因。如DFNA1型耳聋,于1992年定位在5q31,直到1997年,Lynch等发现其致病基因为DIAPH1(diaphanous related formin 1),临床表型为5~30岁发病,听力损失为低频到全频的进展性听力下降。临床上,由DIAPH1基因突变所导致的耳聋可以表述为:DFNA1型耳聋,或者是DIAPH1基因突变型耳聋。
目前已知的常染色体显性遗传性耳聋的基因座位已命名到DFNA67,意为有67个基因座位,但目前发现的致病基因只有32个,还有一半多基因座位上的基因处于尚待发现和进一步确认当中。DFNA1~DFNA67型基因座位及致病基因与临床表型特征见表1,该表的总结可以帮助对显性遗传性耳聋患者的诊断和遗传咨询;在临床上,当疑为常染色体显性遗传性耳聋的家系或患者时,要考虑其是否为DFNA1~DFNA67型耳聋亚型中的一种,或是新的耳聋亚型,应从基因座位、定位区段、致病基因、发病年龄、听力损失的类型、预后、病情是否进展、前庭是否受累等几方面进行诊断和咨询。
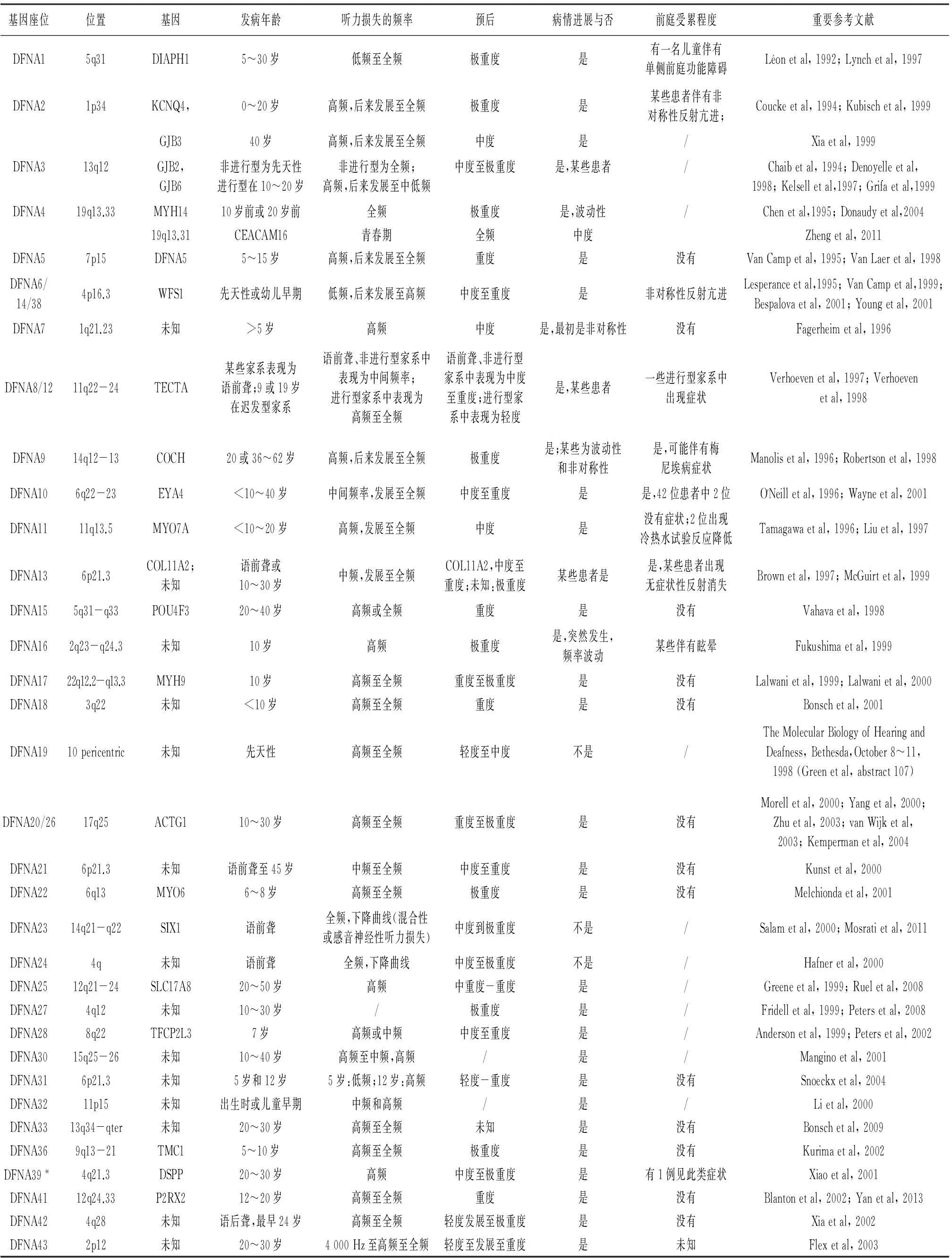
表1 DFNA1~DFNA67型基因座位及致病基因与临床表型特征一览表
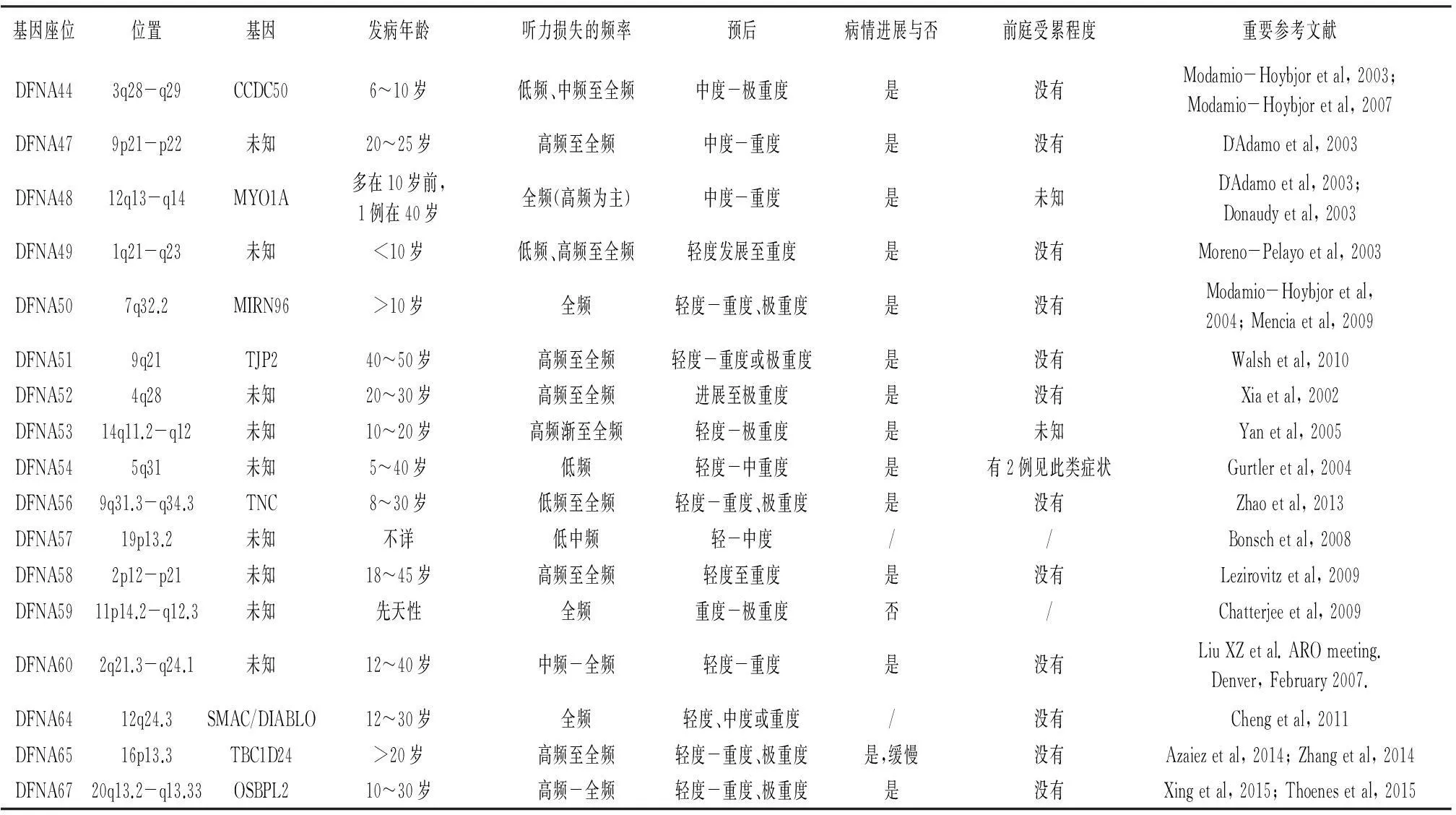
基因座位位置基因发病年龄听力损失的频率预后病情进展与否前庭受累程度重要参考文献DFNA443q28-q29CCDC506~10岁低频、中频至全频中度-极重度是没有Modamio-Hoybjoretal,2003;Modamio-Hoybjoretal,2007DFNA479p21-p22未知20~25岁高频至全频中度-重度是没有D'Adamoetal,2003DFNA4812q13-q14MYO1A多在10岁前,1例在40岁全频(高频为主)中度-重度是未知D'Adamoetal,2003;Donaudyetal,2003DFNA491q21-q23未知<10岁低频、高频至全频轻度发展至重度是没有Moreno-Pelayoetal,2003DFNA507q32.2MIRN96>10岁全频轻度-重度、极重度是没有Modamio-Hoybjoretal,2004;Menciaetal,2009DFNA519q21TJP240~50岁高频至全频轻度-重度或极重度是没有Walshetal,2010DFNA524q28未知20~30岁高频至全频进展至极重度是没有Xiaetal,2002DFNA5314q11.2-q12未知10~20岁高频渐至全频轻度-极重度是未知Yanetal,2005DFNA545q31未知5~40岁低频轻度-中重度是有2例见此类症状Gurtleretal,2004DFNA569q31.3-q34.3TNC8~30岁低频至全频轻度-重度、极重度是没有Zhaoetal,2013DFNA5719p13.2未知不详低中频轻-中度//Bonschetal,2008DFNA582p12-p21未知18~45岁高频至全频轻度至重度是没有Lezirovitzetal,2009DFNA5911p14.2-q12.3未知先天性全频重度-极重度否/Chatterjeeetal,2009DFNA602q21.3-q24.1未知12~40岁中频-全频轻度-重度是没有LiuXZetal.AROmeeting.Denver,February2007.DFNA6412q24.3SMAC/DIABLO12~30岁全频轻度、中度或重度/没有Chengetal,2011DFNA6516p13.3TBC1D24>20岁高频至全频轻度-重度、极重度是,缓慢没有Azaiezetal,2014;Zhangetal,2014DFNA6720q13.2-q13.33OSBPL210~30岁高频-全频轻度-重度、极重度是没有Xingetal,2015;Thoenesetal,2015
注:表中基因座位一栏按基因是否克隆分类排列,其中未包括目前仍保留名称但未确认定位的座位。*DFNA39不是非综合征耳聋表型,在一些家系中表现为牙本质发育不全伴有听力下降
遗传性耳聋的一个显著特征就是遗传的异质性,同样的表型由不同的基因所导致,或者是同样的基因导致不同的耳聋表型。这种遗传的异质性在常染色体显性遗传性耳聋中,又有两个值得关注的特点:一是基因座位与耳聋基因的多关联性;二是致聋基因既表现为显性遗传又表现为隐性遗传的显隐共性特征,表现为如下两个方面:①一个基因座中含两个不同的致聋基因:在目前已经鉴定的32个DFNA耳聋基因中,相对应的基因座位为27个,即并非所有的基因座位都只有一个耳聋基因。从表2可以看到DFNA2、DFNA3、DFNA4基因座位上,均有2个耳聋基因,因此,在遗传性耳聋官方网站上,将其分别列出小亚型,如DFNA2型耳聋基因座位分出了DFNA2A亚型和DFNA2B亚型,对应的致病基因分别为KCNQ4和GJB3;同样DFNA3型耳聋基因座位又分为DFNA3A亚型和DFNA3B亚型,对应的基因分别为GJB2和GJB6;DFNA4型耳聋基因座位是1995年定位的一个显性遗传性耳聋基因座,2004年发现了第一个耳聋基因为MYH14,时隔七年之后的2011年,发现了第二个责任基因CEACAM16,因此,也分为DFNA4A亚型和DFNA4B亚型。
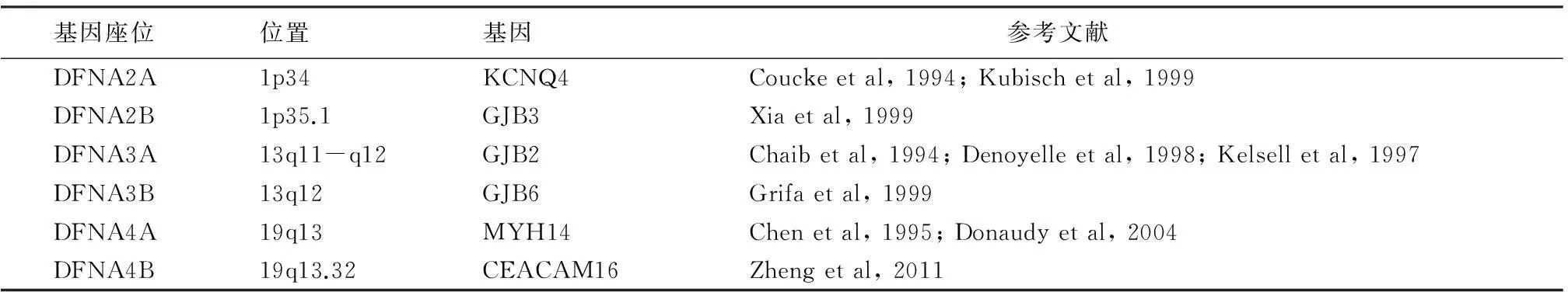
表2 DFNA2、DFNA3、DFNA4各亚型的对应位置及致病基因
②一个基因对应两个或三个基因座位,这些基因既可以表现为显性遗传(DFNA),也可以表现为隐性遗传(DFNB),遗传方式不同临床表型特征亦不同;如:GJB2等6个基因就对应于2个基因座,而TECTA等3个基因则对应于3个基因座(表3),而且这些基因座分别以显性和隐性不同方式遗传。以TECTA基因在DFNA8/DFNA12和DFNB21型耳聋为例:TECTA基因编码的α-tectorin蛋白,其突变引起非综合征型显性DFNA8/DFNA12型和隐性DFNB21型耳聋,DFNA8/DFNA12型听力损失的表型为语前聋,稳定的中频听力损失;该基因的错义突变引起α-tectorin蛋白透明带(zona pellucida)的保守氨基酸残基的替换而影响该蛋白的功能;一个DFNA12型听力损失家系表现的是学语后的、进行性的听力损失,基因突变引起α-tectorin蛋白的zonadhesion/von Wilebrand区域的一个丝氨酸代替了原有的半胱氨酸而引起耳聋。而同样由TECTA基因引起的DFNB21型耳聋表型为语前发病,重度至极重度感音神经性耳聋,TECTA基因的一个剪切突变导致一个α-tectorin截短蛋白的出现而引起听力损失。同为TECTA基因引起表型迥异的原因,目前认为有两种可能的解释:一是同一个基因的不同突变将在不同程度上影响α-tectorin蛋白与其它分子相互作用的能力,并引起非胶原盖膜基质结构的不同程度的改变而导致不同程度的听力损失;另一种可能是由于TECTA基因的修饰基因的作用影响所致。
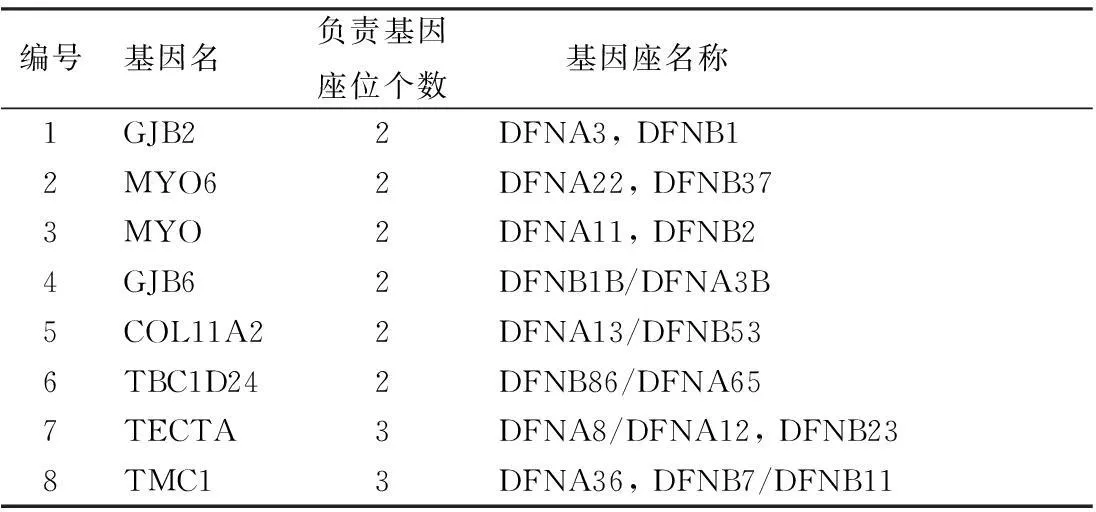
表3 对应于多个基因座位基因的负责基因座位个数及名称
综上所述,本文阐述了常染色体显性遗传性耳聋的致病基因的命名原则、定位方法和基因发现的基础知识,涵盖了一些必须了解的概念,以便理解常染色体显性遗传性耳聋的遗传学特征和临床特征。遗传性耳聋基因的发现是一个巨大的工程,是人类揭秘遗传性耳聋本质,由此能够精准诊断疾病,未来可以实现有效基因治疗的基础。从上述内容中,也可以看到遗传性耳聋的复杂性和多样性,了解一个个基因,认识一个个表型,才能实现临床上的精准诊断。随着新一代测序技术的飞速发展,越来越多的耳聋基因被发现,而其对应的临床特征的解析任务仍然非常复杂而繁重,需要持续的努力。随后的章节,将介绍常染色体显性遗传性耳聋的临床特征与遗传咨询内容。
2参考文献
1http://hereditaryhearingloss.org/ Hereditary Hearing loss Homepage.
2Toriello HV, Smith SD. Hereditary hearing loss and its syndromes (Third edition)[M]. America: Oxford University Press, 2013.105~146.
3王秋菊, 韩东一, 翟所强,等. 遗传性听力损失及其综合征[M].第2版.北京:人民军医出版社, 2006.49~79.
4Leon PE, Raventos H, Lynch E, et al. The gene for an inherited form of deafness maps to chromosome 5q31[J]. Proc Nati Acad Sci USA, 1992, 87:5181.
5Lynch ED, Lee MK, Morrow JE,et al. Nonsyndromic deafness DFNA1 associated with mutation of a human homolog of the Drosophila gene diaphanous[J]. Science, 1997, 278:1315.
(待续)
(2016-02-22收稿)
(本文编辑周涛)
【中图分类号】R764.44
【文献标识码】A
【文章编号】1006-7299(2016)02-0213-04
DOI:10.3969/j.issn.1006-7299.2016.02.027
通讯作者:王秋菊(Email:wqcr@263.net 或wqcr301@sina.com)
·继续教育园地·
*国家重大科学研究计划项目(2014CB943001)、国家自然基金重点国际合作项目(81120108009)、国家自然科学基金重点项目(81530032)联合资助
1解放军总医院解放军耳鼻咽喉研究所 (北京100853)
猜你喜欢
中国现代医生(2022年19期)2022-11-04
昆明医科大学学报(2022年4期)2022-05-23
中国听力语言康复科学杂志(2021年6期)2021-12-21
中医眼耳鼻喉杂志(2021年1期)2021-07-22
中国生殖健康(2020年4期)2021-01-18
临床肝胆病杂志(2020年1期)2020-12-20
中国生殖健康(2018年4期)2018-11-06
消费导刊(2017年24期)2018-01-31
中国科技纵横(2016年15期)2016-12-29
中学语文(2015年27期)2015-03-01
