
基于Miseq的好氧反硝化菌源水脱氮的种群演变
2016-05-27 07:32周石磊黄廷林张春华白士远何秀秀西安建筑科技大学环境与市政工程学院西北水资源与环境生态教育部重点实验室陕西西安710055
中国环境科学 2016年4期
周石磊,黄廷林,张春华,白士远,何秀秀 (西安建筑科技大学环境与市政工程学院,西北水资源与环境生态教育部重点实验室,陕西 西安 710055)
基于Miseq的好氧反硝化菌源水脱氮的种群演变
周石磊,黄廷林*,张春华,白士远,何秀秀 (西安建筑科技大学环境与市政工程学院,西北水资源与环境生态教育部重点实验室,陕西 西安 710055)
摘要:为了研究好氧反硝化菌源水脱氮过程中水体微生物群落的演变,利用Miseq高通量测序法对投菌和对照两系统水体样本的微生物信息进行统计,并对两组样品进行了优化序列统计,OTU分布统计和分类学分析的基础分析;以及细菌群落结构,PCA,Rank-Abundance, Hcluster, Specaccum和OTU分布的高级分析.结果显示,投加贫营养好氧反硝化菌的源水系统的氮素得到有效去除,脱氮效果明显;层次聚类和主成分分析显示两系统内的群落结构发生变化,投菌系统与对照系统主要表现为变形菌和拟杆菌门;细菌主要门类和水质参数的相关性分析得出,水质指标对两系统群落变化作用明显;与此同时,投菌系统中有关氮循环的细菌有上升的变化过程. Miseq高通量测序研究源水脱氮过程的微生物种群演变可行,为研究原位生物脱氮过程的水体微生物群落演变提供技术支撑.
关键词:源水脱氮;好氧反硝化;Miseq测序;生物信息分析;微生物群落
∗ 责任作者, 教授, huangtinglin@xauat.edu.cn
20世纪80年代,Robertson等[1]在除硫和反硝化处理系统中首次分离了好氧反硝化菌(Thiosphaera pantotropha),这种新型脱氮生物的发现使人们对氮循环又有了新的认识.好氧反硝化菌[2]是在有氧条件下,利用好氧反硝化酶进行反硝化作用的一类脱氮菌.它的发现打破了反硝化只能在厌氧缺氧条件下进行的传统反硝化的观念,为新型生物脱氮提供了新思路.目前所分离的好氧反硝化菌主要分布于假单胞菌属(Pseudomonas)[3]、芽孢杆菌属(Bacillus)[4-5]、产碱菌属(Alcaligenes)[6-7]、微枝杆菌属(Microvirgula)[8-9]和丛毛单胞菌属(Comamonas)[10-11].目前大多数关于好氧反硝化菌的研究集中在实验室富营养配水培养条件下以及污废水条件下的脱氮特性分析.如, 张培玉等[12]在富营养条件下(初始氮素109mg/L,COD 约2000mg/L)研究好氧反硝化菌假单胞菌qy37的脱氮特性;于爱茸等[13]分离的芽孢杆菌W2具有独特的好氧反硝化作用,0.067hm2鱼塘投入1Kg的该菌就能完全解决除氮问题;Joo等[14]使用Alcaligenes faecalis 处理猪场废水,总氮去除率高于65%;Bouchez等[15]将Microvirgula aerodenitrificans包埋于藻朊酸盐中,投加于传统硝化反应器中,用于市政废水处理,HN-AD脱氮率达26.8%;汪苹等[16]在高浓度硝氮的富营养培养基条件下研究丛毛单胞菌WXZ-17的反硝化特性,初始硝氮为92mg/L培养4d后硝氮去除率达到61%.
然而,针对微污染水源水体的贫营养好氧反硝化菌的分离鉴定及其脱氮特性的研究很少.对于好氧反硝化菌进行源水脱氮过程中,微生物种群的演变更鲜有报道.1979年Kuznetsov等[17]提出寡营养细菌的概念,并定义第一次培养时能在含碳1~15mg/L培养基中生长的细菌称为寡营养细菌,能同时在贫营养和富营养基中生长的称为兼性寡营养细菌.Wainwright等[18]和Button等[19]研究表明,贫营养细菌对营养物质碳的亲和力较大,在低营养环境中,具有较大的营养竞争优势.结合本课题组之前关于贫营养好氧反硝化菌的研究[20-23],通过从微污染水体水库底层沉积物中富集、驯化、分离出一批贫营养好氧反硝化菌,并通过对这些贫营养好氧反硝化菌进行源水梯度驯化,使其适应贫营养环境. 源水驯化完成后接种到微污染源水环境下,考察其在低氮微污染水体下的脱氮特性.
近年来,应用分子生物学手段来研究微生物群落结构变化成为热点,如白玉涛等[24]应用末端限制性片段技术对反硝化和甲烷氧化细菌的群落进行分析;党岩等[25]应用克隆文库对应用EGSB 处理垃圾焚烧渗沥液的微生物群落变化进行研究;郭建宁等[26]应用HiSeq 2000建Illumina测序文库对臭氧/陶瓷膜对生物活性炭工艺的微生物群落结构进行分析.第2代高通量测序技术Illumina MiSeq以Illumina的边合成边测序技术为基础,具有快速准确的分析特点,被广泛应用于微生物群落结构及多样性分析[27-28].结合该源水投菌实验已发表的关于氮素以及有机物变化特性[29],本文主要针对在静态源水条件下,应用Miseq高通量测序技术对两系统的起始、过程以及结束3个阶段的水样进行分析,来考察投菌系统在源水脱氮过程中的微生物群落演变过程.为以后分析原位生物强化水源水体过程中对水体微生物群落影响提供技术支撑.
1 材料与方法
1.1 实验装置
实验装置为20L的玻璃瓶,外壁附有黑色塑料袋来模拟水库的黑暗环境.取山东周村水库源水(2013年8月)并通过充氧泵的间歇曝气来控制反应器的溶解氧,实验的温度为室温.经梯度驯化好的三株贫营养好氧反硝化菌Zoogloea sp. N299 (GenBank登录号,KP717093)[29], Acinetobacter sp. G107 (GenBank登录号, KP717096)[29]和Acinetobacter sp. 81Y (GenBank登录号,KP717097)[29]分别以体积比2mL/20L.活化后菌液的好氧反硝化菌的菌密度达到106cfu/mL.通过测定反应器水样中NO3--N, NO2--N,TN,COD和好氧反硝化菌菌落数来反应投菌的源水实验效果. NO3--N,NO2--N和TN每5d一测;菌落数为10d一测.实验装置示意如图1所示.

图1 源水实验装置示意Fig.1 The schematic diagram of experiment system
1.2 微生物多样性分析
1.2.1 DNA提取与PCR扩增 在进行投加贫营养好氧反硝化菌源水生物脱氮实验的初始阶段(0d),中间阶段(30d),结束阶段(60d).共3次从投菌系统和对照系统分别取1L的水样经0.22μm醋酸纤维滤膜过滤,以备提取DNA.将投菌系统和对照系统的水样滤膜利用水样DNA提取试剂盒完成基因组DNA的抽提(美吉生物,上海).抽提DNA的含量和质量采用分光光度法进行检验.PCR扩增区域选择样本16S rRNA基因的V1~V3区域,合成带有barcode的特异引物.全部样本按照正式实验条件进行,每个样本3个重复,将同一样本的PCR产物混合后用2%琼脂糖凝胶电泳检测,使用AxyPrepDNA凝胶回收试剂盒(AXYGEN公司)切胶回收PCR产物,Tris_HCl洗脱; 2%琼脂糖电泳检测.参照电泳初步定量结果, 将PCR产物用QuantiFluorTM-ST蓝色荧光定量系统(Promega公司)进行检测定量,之后按照每个样本的测序量要求,进行相应比例的混合.
1.2.2 Miseq文库构建和测序 应用新一代高通量测序平台Miseq对扩增产物进行测序.Miseq文库构建: (1)连接“Y”字形接头; (2)使用磁珠筛选去除接头自连片段; (3)利用PCR扩增进行文库模板的富集; (4)氢氧化钠变性,产生单链DNA片段. Miseq测序:(1)DNA片段的一端与引物碱基互补,固定在芯片上; (2)另一端随机与附近的另外一个引物互补,也被固定住,形成“桥 (bridge)” ; (3)PCR扩增,产生DNA簇; (4)DNA扩增子线性化成为单链; (5)加入改造过的DNA聚合酶和带有4种荧光标记的dNTP,每次循环只合成一个碱基; (6)用激光扫描反应板表面,读取每条模板序列第一轮反应所聚合上去的核苷酸种类; (7)将“荧光基团”和“终止基团”化学切割,恢复3'端粘性,继续聚合第二个核苷酸; 统计每轮收集到的荧光信号结果,获知模板DNA片段的序列.
1.2.3 生物多样性与分类学分析 将Miseq测序得到的PE reads首先根据overlap关系进行拼接,同时对序列质量进行质控和过滤,区分样本后进行OTU聚类分析和物种分类学分析,基于OTU可以进行多种多样性指数分析[30],基于OTU聚类分析结果,可以对OTU进行多种多样性指数分析,以及对测序深度的检测;基于分类学信息,可以在各个分类水平上进行群落结构的统计分析[31].在上述分析的基础上,可以进行一系列群落结构和系统发育等深入的统计学和可视化分析[30,32-33].
1.3 水质分析方法
水质指标NO3--N,NO2--N和TN采用分光光度计测定[34].具体如下, NO3--N采用紫外分光光度法, NO2--N为N-(1-奈基)-乙二胺光度法, TN为过硫酸钾氧化-紫外分光光度法.好氧反硝化菌菌落数采用平板计数法[29,35].NO3--N, NO2--N的水样预处理为进过0.45μm醋酸纤维滤膜过滤.醋酸纤维滤膜经过超纯水的3次煮沸,每次沸腾半小时去除杂质.
2 结果与讨论
2.1 源水脱氮效果分析
结合之前研究结果[29]如下图2所示,在温度为20~27℃,DO在3~7mg/L条件下,通过60d的源水实验,投菌系统的NO3--N从初始的(1.57± 0.02)mg/L下降到(0.42±0.01)mg/L,对照系统的NO3--N由(1.63±0.02)mg/L下降到(1.30± 0.01)mg/L;两系统都没有NO2--N的积累;投菌系统的TN从初始(2.31±0.12)mg/L下降到(1.09± 0.01)mg/L,对照系统的TN从(2.51±0.13)mg/L下降到(1.72±0.06) mg/L;投菌系统的好氧反硝化菌菌落数从初始的2.8×104上升到2×107cfu/mL,对照系统从7.75×103到5.5×105cfu/mL.结果表明投加好氧反硝化菌可以实现源水脱氮.
2.2 微生物多样性分析
通过对投菌和对照两系统3个时期取样的样品16S rRNA基因进行测序,在实验初始阶段投菌系统样品和对照系统样品各获得了22412 和19776条有效序列,序列平均长度315bp;中间过渡阶段投菌系统样品和对照系统样品各获得了21433和27171条有效序列,序列平均长度318bp;结束阶段投菌系统样品和对照系统样品各获得了25475和16483条有效序列,序列平均长度317bp.通过Rank-abundance曲线(等级-多度曲线)[36],考察源水投菌实验过程中细菌的物种丰度和物种均匀度.

图2 源水实验中氮素及好氧反硝化菌的变化情况Fig.2 The changes of nitrogen concentrations and aerobic denitrification bacteria in source water experiment
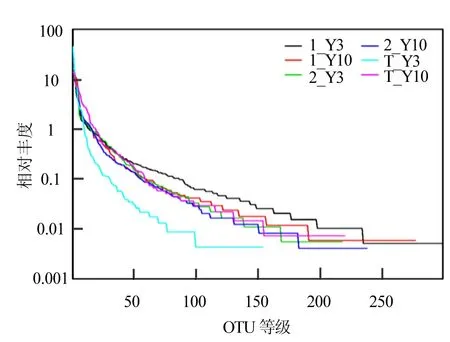
图3 源水实验的等级-多度曲线Fig.3 The rank-abundance curve of source water experiment
图3中展示的为源水实验过程中投菌系统和对照系统的Rank-abundance的变化情况.在水平方向,曲线的宽度来反映物种的丰富度,宽度越大表示丰富度越高;曲线形状的平滑程度反映样本中物种的均度,曲线越平缓说明物种分布越均匀.投菌系统和对照系统从源水实验的开始阶段到结束阶段,细菌种群的丰富度逐渐减小.然而,源水投菌系统的Rank-abundance逐渐变陡峭,表示物种均度随时间变得不均匀;对照系统的物种均度变化不明显.
2.3 细菌群落结构组分
将源水实验的投菌系统和对照系统共6个点的样本,共116572个OTU进行注释,然后统计在属类别上的构成形成柱状图(图4),同时分析在各个水平上的菌群结构.结果显示这116572个 OTU主要属于7个门类44个属,具体结果如图4和表1所示.

图4 源水实验的细菌群落结构组分Fig.4 The bacterial community during the water of bacteria and control experimental systems
两系统的OUT主要属于7个门类,分别是变形菌门(Protebacterice)、拟杆菌门(Bacteroidetes)、放线菌门(Actinobacteria)、蓝藻门(Cyanobacteria)、酸杆菌门(Acidobacteria)、装甲菌门(Armatimonadetes)和其他少数细菌门类,其中主要的细菌门类为变形菌门.对照系统和投菌系统的变形菌门从开始的32.83%和39.02%上升到实验中间阶段的87.55%和86.24%,到实验结束时变为93.05%和94.97%.结果显示,在源水脱氮过程中,对照和投菌两系统的细菌菌落在门的类别上变化相差不大.在变形菌门类中对照和投菌两系统在α-变形菌和β-变形菌变化较大.其中,对照系统中α-变形菌呈现上升趋势,从开始阶段的12.48%到中间阶段的18.27%,最后到31.05%;投菌系统中α-变形菌呈现下降趋势,从开始阶段的19.12%到中间阶段的11.16%,最后到7.56%.而两系统中β-变形菌都是上升趋势,对照和投菌系统从开始的19.20%和18.53%上升到实验中间阶段的49.80%和86.39%.其他类的变形菌变化不大.
在源水脱氮实验过程中,对照和投菌两系统的细菌群落分析得出44个属. 投菌源水实验系统的开始阶段主要类别如下, SubsectionIII_ FamilyI_unclassified (32.74%)、LD12_freshwater_ group_norank (14.52%)、Azospira (8.43%)、CL500-29_marine_group(5.85%)、Polynucleobacter (3.81%)、hgcI_clade (2.52%)、CL500-3 (2.23%);中间阶段时期主要类别如下, Hydrogenophaga (42.70%)、Betaproteobacteria_unclassified (10.56%)、Comamonadaceae_unclassified (8.26%)、NS11-12_marine_group_norank (7.53%)、Limnohabitans (4.20%)、Novosphingobium (3.35%);结束阶段时期主要类别如下, Limnohabitans (59.62%)、Aquincola (19.78%)、Reyranella (5.92%)、Comamonadaceae_ unclassified (4.49%).
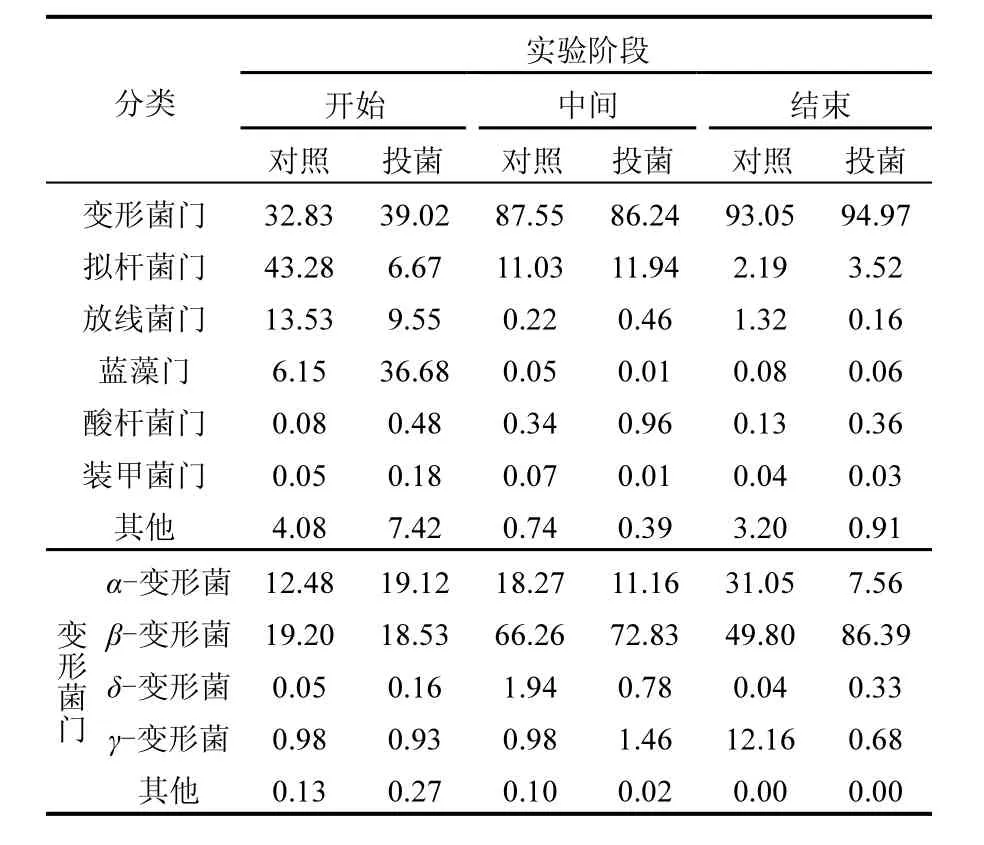
表1 源水实验中细菌菌落结构变化情况(%)Table 1 The bacterial community during the source water experiment (%)
对照系统开始阶段主要类别如下, Flavobacterium (40.36%)、Azospira (11.23%)、LD12_freshwater_group_norank (10.31%)、CL500-29_marine_group (9.40%)、SubsectionIII_ FamilyI_unclassified (5.03%)、hgcI_clade (2.96%)、Polynucleobacter (2.43%);中间阶段时期主要类别如下, Comamonadaceae_unclassified (40.89%)、Aquincola (17.06%)、Reyranella (8.59%)、Candidatus_Aquirestis (4.22%)、 Sphingomonadaceae_unclassified (3.54%)、Betaproteobacteria_unclassified (3.36%);结束阶段时期主要类别如下, Limnohabitans (22.71%)、Methyloversatilis (12.63%)、Sphingomonadaceae_ unclassified (10.38%)、Aquabacter (6.76%)、Reyranella (5.71%)、Acinetobacter (4.74%)、Stenotrophomonas (4.69%)、Betaproteobacteria_ unclassified (4.66%)、Comamonadaceae_ unclassified (3.34%)、Hydrogenophaga (2.76%).
结果显示,与氮循环相关的β变形菌(Betaproteobacteria,如Limnohabitans和Acidovorax)[37]:投菌系统中从开始阶段的0.02%,上升到中间阶段的4.32%,到最后结束阶段的60.19%;与此同时,对照系统的Limnohabitans从开始阶段的0.05%,上升到中间阶段的0.84%,到最后阶段的23.53%.投菌系统的Acidovorax从开始的0.11%上升到实验结束的0.22%;对照系统基本不存在.另一种菌为代尔夫特菌 (Delftia)与我们课题组之前从原水中分离的一株贫营养好氧反硝化菌SF9属于同一种属:投菌系统中从开始阶段的0.65%,上升到中间阶段的8.50%,最后到结束阶段的4.53%;对照系统的从开始的0.38%,上升到中间阶段的42.02%,最后到结束阶段的3.46%.另外一β变形菌 (Aquincola)[38]:投菌系统中从开始阶段的0.01%,上升到中间阶段的1.55%,到最后结束阶段的19.97%;与此同时,对照系统的从开始阶段的0%,上升到中间阶段的17.53%,到最后阶段的1.34%.赵文莉等[28]研究复合填料反硝化脱氮过程的微生物群落特征时,氢噬胞菌(Hydrogenophaga)具有反硝化功能,本实验中:投菌系统中从开始阶段的0.02%,上升到中间阶段的43.94%,到最后结束阶段的0.73%;与此同时,对照系统的从开始阶段的0.01%,上升到中间阶段的2.03%,到最后阶段的2.86%.不动杆菌 (Acinetobacter)与源水所投加的81Y和G107所属种类一致:投菌系统中从开始阶段的0.10%,上升到中间阶段0.59%,到最后结束阶段的0.00%;与此同时, 对照系统的从开始阶段的0.01%,上升到中间阶段的0.15%,到最后阶段的4.91%.与此同时,主要门类细菌与水质参数的相关性分析如表2所示,结果显示,投菌和对照系统在变形菌门的变化与系统的氮素参数呈现明显的负相关,但是在拟杆菌门细菌与水质参数相关关系存在明显差异,对照系统的正相关性更强.体现出两系统细菌群落变化的差异性.相关性显示,氮素去除主要表现为硝氮的下降.虽然所投加的好氧反硝化菌并没有在细菌群落的heatmap图发生显著的增加,可能是由于反硝化菌的投加与水体中存在的氮循环菌发生协同作用,促进了其他好氧反硝化菌的脱氮进行.
本实验中对照和投菌两系统的变形菌是重要变化的细菌门类,并且变化趋势一致.与此同时,投菌系统水体中的反硝化菌菌落数目在增加.通过Heatmap得出,具有反硝化功能的细菌,其中β变形菌(Limnohabitans, Aquincola和Acidovorax)、代尔夫特菌(Delftia)、氢噬胞菌(Hydrogenophaga)以及不动杆菌(Acinetobacter)的丰度随实验进行获得提高.

表2 源水实验中主要细菌群落与水质参数的相关关系Table 2 The correlation analysis between main bacterial community and water parameters in bacteria and control systems
Meta-群落假说[39-40]认为,细菌多样性是因扩散作用而联系在一起的一系列本地群落的集合,它强调不同时空尺度上群落的结构和动态过程.在meta-群落假说中,有两个影响因子影响细菌群落组成:内部环境因子和外部环境因子.其中内部环境因子指食物网中生物间的相互作用及物种的随机动态等物种本身之间的影响因子,而外部环境因子指环境温度、主要离子浓度等外在因素.目前一般认为在水力停留时间长的水体(如湖泊和水库)中细菌群落主要受其内部环境因子的影响[40].但是,本文仅仅考察了在静态源水系统中投加贫营养好氧反硝化菌,脱氮过程中水体的细菌群落以及多样性的演变过程,存在一定的局限性.考虑以后增加源水动态过程以及底泥存在条件下脱氮变化中的细菌群落变化特征的考察,以期为微污染水源水库的生物修复过程中对水体细菌群落的影响提供技术支持.
2.4 源水实验的细菌群落主成分分析
将源水实验投菌和对照两系统的开始时期、中间时期和结束时期共6个样点的OTU (97%相似性)进行主成分分析,来反映样本间的差异和距离.样本间的组成越相似,反映在PCA图中的距离越近.通过在PCA图中的样本间的分散和聚集分布反映样本间细菌群落的相似程度.图5的结果展示,6个样点的主成分分析得出PC1+PC2达到68.38%.在源水实验开始阶段,投菌系统和对照系统的细菌群落组成相似性很高;随着源水实验的进行,各个系统的细菌群落组成发生变化,同时期的投菌系统与对照系统的细菌群落差异很大.与两系统的OTU分布的Veen图特征和样本间聚类特征相一致.

图5 源水实验的细菌主成分分析Fig.5 Principal component analysis (PCA) of bacteria and control source experimental systems
2.5 源水实验的OTU分布Venn图特征
Venn图可用于统计多个样本中所共有和独有的OTU数目,可以比较直观的表现环境样本的OTU数目组成相似性及重叠情况. 本实验选用相似水平为97%的OTU样本表.
投菌系统和对照系统的结果如图6所示.源水实验投菌系统结果显示为,开始阶段与中间阶段共有85个相同的OTU样本;中间阶段与结束阶段共有102个相同的OTU样本;开始阶段与结束阶段共有54个相同的OTU样本;开始阶段、中间阶段与结束阶段共有45个相同的OTU样本.源水实验对照系统结果显示为,开始阶段与中间阶段共有70个相同的OTU样本;中间阶段与结束阶段共有143个相同的OTU样本;开始阶段与结束阶段共有61个相同的OTU样本;开始阶段、中间阶段与结束阶段共有49个相同的OTU样本,对照系统三阶段共有的OTUs要高于投菌系统.

图6 源水实验两系统的OUT分布Fig.6 OUT distribution of bacteria and control systems in source water experiment
然而,投菌系统和对照系统间的菌群间相似性如图7所示,开始阶段时投菌系统 对照系统共有212个相同的OTU样本;中间阶段时投菌系统对照系统共有158个相同的OTU样本;结束阶段时投菌系统和对照系统共有95个相同的OTU样本. 随着源水实验的进行同一系统中从开始到结束阶段,投菌系统内共有的OTUs要小于对照系统,并且同一时期投菌系统与对照系统共有的OTUs也随着试验的进行逐渐减少.从源水实验开始到结束整个实验过程中两系统共有的OTUs明显越来越少,说明投菌系统和对照系统间的细菌群落差异性逐渐显现,这点与6个样本间的聚类分析相一致.

图7 源水实验不同时期的OUT分布Fig.7 OUT distribution of different periods in source experiment
2.6 源水实验的样本间聚类特征
利用树枝结构描述和比较多个样本间的相似性和差异关系.首先使用描述群落组成关系和结构的算法计算样本间的距离,即根据beta多样性距离矩阵进行层次聚类分析,使用非加权组平均法UPGMA(Unweighted pair group method with arithmetic mean)算法构建树状结构,得到树状关系形式用于可视化分析.通过bray curtis算法使用qiime计算beta多样性距离矩阵,算法如下:

式中:SA,i= 表示A样本中第i个OTU所含的序列数; SB,i= 表示B样本中第i个OTU所含的序列数.
本源水实验的投菌系统和对照系统从实验开始阶段、中间阶段和结束阶段取的6个样本,通过样本的OTUs进行层次聚类分析得到结果如图8所示.
结果显示,实验初始阶段投菌系统和对照系统在同一分支下,样本间相似性很高、差异不明显;随着源水实验的进行,投菌系统和对照系统内样本间的OTUs被分离,两系统内的细菌群落结构发生变化.在贫营养好氧反硝化菌作用下的投菌系统中,随着实验的进行系统内水体的氮素在逐渐减小.因此水体的微生物生存受到限制,其物种丰富度下降,与细菌门类与对照和投菌系统水质参数相关性分析结果相一致.
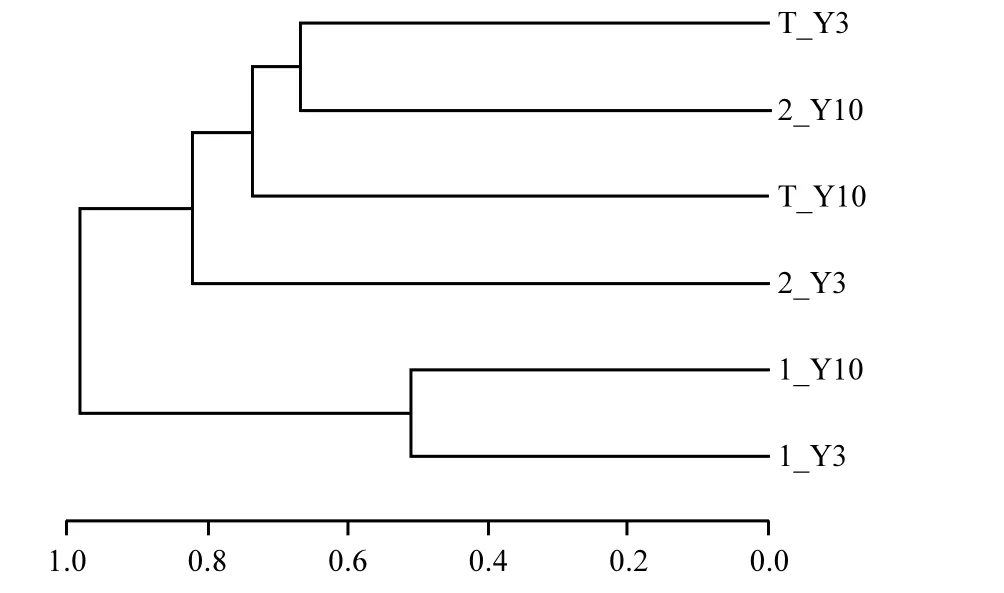
图8 源水实验细菌多样本相似度树状图Fig.8 The hcluster tree of source water experimental system
3 结论
基于Miseq高通量测序法研究源水脱氮过程的微生物群落的演变.结果显示投菌系统与对照系统的水体中的微生物群落结构随着实验进行都发生了变化,两系统不同时期差异性明显.其中两系统中变形菌的群落结构与氮素指标存在明显的负相关.与此同时投菌系统中与氮循化相关的菌种明显增加.考虑到静态源水投菌实验的局限性,以后增加底泥存在下流动源水投菌实验的细菌群落变化,以期为研究微污染水源水库的投加贫营养好氧反硝化菌进行生物修复过程中对水体细菌群落的影响提供技术支持.
参考文献:
[1] Robertson L A, Kuenen J G. Thiosphaera pantotropha gen. nov. sp. nov., a facultatively anaerobic, facultatively autotrophic sulphur bacterium [J]. Journal of General Microbiology, 1983, 129(9):2847-2855.
[2] Gao H, Schreiber F, Collins G, et al. Aerobic denitrification in permeable Wadden Sea sediments [J]. The ISME journal, 2010, 4(3):417-426.
[3] Zhang J B, Wu P X, Hao B, et al. Heterotrophic nitrification and aerobic denitrification by the bacterium Pseudomonas stutzeri YZN-001 [J]. Bioresource Technology, 2011,102(21):9866-9869.
[4] Yang X P, Wang S M, Zhang D W, et al. Isolation and nitrogen removal characteristics of an aerobic heterotrophic nitrifying–denitrifying bacterium, Bacillus subtilis A1 [J]. Bioresource Technology, 2011,102(2):854-862.
[5] Yu A, Li Y, Yu J. Denitrification of a newly isolated Bacillus strain W2and its application in aquaculture [J]. Journal of Microbiology, 2004,25(3):77-81.
[6] Zhao B, An Q, He Y L, et al.N2O and N2production during heterotrophic nitrification by Alcaligenes faecalis strain NR [J]. Bioresource Technology, 2012,116(0):379-385.
[7] Joo H S, Hirai M, Shoda M. Characteristics of ammonium removal by heterotrophic nitrification-aerobic denitrification by Alcaligenes faecalis No. 4 [J]. Journal of Bioscience and Bioengineering, 2005,100(2):184-191.
[8] Cleenwerck I, De Wachter M, Hoste B, et al. Aquaspirillum dispar Hylemon et al. 1973 and Microvirgula aerodenitrificans Patureau et al. 1998 are subjective synonyms [J]. International Journal of Systematic and Evolutionary Microbiology, 2003, 53:(1457-1459).
[9] Patureau D, Helloin E, Rustrian E, et al. Combined phosphate and nitrogen removal in a sequencing batch reactor using the aerobic denitrifier, Microvirgula aerodenitrificans [J]. Water Research, 2001,35(1):189-197.
[10] Chen Q, Ni J R. Heterotrophic nitrification–aerobic denitrification by novel isolated bacteria [J]. J. Ind. Microbiol. Biotechnol., 2011, 38(9):1305-1310.
[11] Patureau D, Bernet N, Moletta R. Combined nitrification and denitrification in a single aerated reactor using the aerobic denitrifier Commonas sp. strain SGLY2 [J]. Water Research, 1997, 31(6):1363-1370.
[12] 张培玉,曲 洋,于德爽,等.菌株qy37的异养硝化/好氧反硝化机制比较及氨氮加速降解特性研究 [J]. 环境科学, 2010,31(8): 1819-1826.
[13] 于爱茸,李 尤,俞吉安.一株耐氧反硝化细菌的筛选及脱氮特性研究 [J]. 微生物学杂志, 2005,25(3):77-81.
[14] Joo H-S, Hirai M, Shoda M. Piggery wastewater treatment using Alcaligenes faecalis strain No. 4with heterotrophic nitrification and aerobic denitrification [J]. Water Research, 2006,40(16): 3029-3036.
[15] Bouchez T, Patureau D, Delgen S J, et al. Successful bacterial incorporation into activated sludge flocs using alginate [J]. Bioresource Technology, 2009,100(2):1031-1032.
[16] Wang P, Li X, Xiang M, et al. Characterization of efficient aerobic denitrifiers isolated from two different sequencing batch reactors by 16S-rRNA analysis [J]. Journal of Bioscience and Bioengineering, 2007,103(6):563-567.
[17] Kuznetsov S, Dubinina G, Lapteva N. Biology of oligotrophic bacteria [J]. Annual Reviews in Microbiology, 1979,33(1):377-387.
[18] Wainaright M, Barakah F, Al-Turk I, et al. Oligotrophic micro-organisms in industry, medicine and the environment [J]. Science Progress, 1990,75(298Pt 3-4):313-322.
[19] Button D, Schut F, Quang P, et al. Viability and isolation of marine bacteria by dilution culture: theory, procedures, and initial results [J]. Applied and Environmental Microbiology, 1993,59(3): 881-891.
[20] 黄廷林,周 娜,张海涵,等.3株贫营养好氧反硝化细菌的分离鉴定及反硝化特性 [J]. 环境工程学报, 2014,8(12):5507-5513.
[21] 黄廷林,魏 巍,王春燕,等.扬水曝气-贫营养生物膜组合技术净化微污染原水中试研究 [J]. 重庆大学学报, 2012,(1):125-131,146.
[22] 魏 巍,黄廷林,苏俊峰,等.1株贫营养好氧反硝化菌的分离鉴定及其脱氮特性 [J]. 生态环境学报, 2010,19(9):2166-2171.
[23] 黄廷林,苏俊峰,李 倩.好氧反硝化菌株的筛选培养及其反硝化性能研究 [J]. 西安建筑科技大学学报:自然科学版, 2009,(5):704-707.
[24] 白玉涛,周 玉,赵 吉.内蒙古高原干涸湖泊反硝化及甲烷氧化细菌的群落分析 [J]. 中国环境科学, 2012,32(7):1293-1301. [25] 党 岩,张 瑞,叶杰旭,等.EGSB处理垃圾焚烧渗沥液及其微生物群落变化 [J]. 中国环境科学, 2013,33(6):999-1004.
[26] 郭建宁,陈 磊,张锡辉,等.臭氧/陶瓷膜对生物活性炭工艺性能和微生物群落结构影响 [J]. 中国环境科学, 2014,34(3):697-704.
[27] 郑林雪,李 军,胡家玮,等.同步硝化反硝化系统中反硝化细菌多样性研究 [J]. 中国环境科学, 2015,35(1):116-21.
[28] 赵文莉,郝瑞霞,王润众,等.复合碳源填料反硝化脱氮及微生物群落特性 [J]. 中国环境科学, 2015,35(10):3003-3009.
[29] Huang T L, Zhou S L, Zhang H H, et al. Nitrogen removal from micro-polluted reservoir water by indigenous aerobic denitrifiers [J]. International Journal of Molecular Sciences, 2015,16(4): 8008-8026.
[30] Wang Y, Sheng H F, He Y, et al. Comparison of the levels of bacterial diversity in freshwater, intertidal wetland, and marine sediments by using millions of illumina tags [J]. Applied and environmental microbiology, 2012,78(23):8264-8271.
[31] Oberauner L, Zachow C, Lackner S, et al. The ignored diversity: complex bacterial communities in intensive care units revealed by 16S pyrosequencing [J]. Scientific Reports, 2013,3:1413.
[32] Fouts D E, Szpakowski S, Purushe J, et al. Next generation sequencing to define prokaryotic and fungal diversity in the bovine rumen [J]. PLOS One, 2012,7(11):e48289.
[33] Jami E, Israel A, Kotser A, et al. Exploring the bovine rumen bacterial community from birth to adulthood [J]. The ISME journal, 2013,7(6):1069-1079.
[34] 国家环境保护总局.《水和废水监测分析方法》.第4版 [M]. 北京:中国环境科学出版社, 2002.
[35] Huang T L, Wei W, Su J F, et al. Denitrification performance and microbial community structure of a combined WLA–OBCO system [J]. PlOS One, 2012,7(11):e48339.
[36] Bates S T, Clemente J C, Flores G E, et al. Global biogeography of highly diverse protistan communities in soil [J]. The ISME journal, 2013,7(3):652-659.
[37] Yan Q, Bi Y, Deng Y, et al. Impacts of the Three Gorges Dam on microbial structure and potential function [J]. Scientific Reports, 2015,5:8605.
[38] Yang X, Xie P, Ma Z, et al. Decrease of NH4+-N by bacterioplankton accelerated the removal of cyanobacterial blooms in aerated aquatic ecosystem [J]. Journal of Environmental Sciences, 2013,25(11):2223-2228.
[39] Leibold M A, Holyoak M, Mouquet N, et al. The metacommunity concept: a framework for mult-scale community ecology [J]. Ecology Letters, 2004,7(7):601-613.
[40] Brendan Logue J, Lindström E S. Biogeography of bacterioplankton in inland waters [J]. Freshwater Reviews, 2008,1(1):99-114.
Bacterial community structures of aerobic denitrification bacteria nitrogen removal process in source water experiment by using Miseq high-throughput sequencing technique.
ZHOU Shi-lei, HUANG Ting-lin*, ZHANG Chun-hua, BAI Shi-yuan, HE Xiu-xiu (Key Laboratory of Northwest Water Resource, Environment and Ecology, Ministry of Education, School of Environmental and Municipal Engineering, Xi’an University of Architecture and Technology, Xi’an 710055, China). China Environmental Science, 2016,36(4):1125~1135
Abstract:Miseq high-throughput sequencing technique was used to investage the bacterial community structure of nitrogen removal process in source water experiment systems added aerobic denitrification bacteria. Bioinformatics analysis were carried on the for the samples of bacteria and control systems. Then, the fundamental analysis, Optimized sequence statistics, OTU distribution, and Taxonomic analysis; the Advanced analysis, the microbial community, PCA, Rank-Abundance, Hcluster, Specaccum, and Venn of OTU distribution. As a result, the nitrogen of bacteria system was removed obviously, and Hierarchical cluatering and PCA showed that the microbial community structure of bacteria and control systems has changed, and the Protebacterice and Bacteroidetes were the main phylum in experiment systems. Meanwhile, the N-functional microbial of bacteria system had an increase process, obviously, which was consistent with the changes of aerobic denitrification bacteria. From all the results, the Miseq high-throughput sequencing technique was an effective tool to explore the changes of bacterial community structure of nitrogen removal process, which could supply a reference to study the changes of community structure of remediation the source water in situ.
Key words:nitrogen removal;aerobic denitrification;Miseq sequencing;bioinformatics analysis;bacterial community structure
作者简介:周石磊(1987-),男,河北石家庄人,西安建筑科技大学博士研究生,主要从事水环境修复与水源水质微污染控制的相关研究.
基金项目:国家科技支撑计划(2012BAC04B02)
收稿日期:2015-09-15
中图分类号:X172
文献标识码:A
文章编号:1000-6923(2016)04-1125-11
