
基于SVM的肿瘤特征基因提取与基因表达数据分析
2016-07-15 03:51王琦然王学敏邱文莹
重庆理工大学学报(自然科学) 2016年6期
谭 云,于 彬,王琦然,王学敏,李 珊,邱文莹
(1.青岛职业技术学院 生物与化工学院,青岛 266555;2.青岛科技大学 数理学院,青岛 266061)
基于SVM的肿瘤特征基因提取与基因表达数据分析
谭云1,于彬2,王琦然2,王学敏2,李珊2,邱文莹2
(1.青岛职业技术学院 生物与化工学院,青岛266555;2.青岛科技大学 数理学院,青岛266061)
摘要:提出一种基于支持向量机的肿瘤基因表达谱数据挖掘方法。首先采用信噪比方法对白血病、结肠癌、肺癌数据提取特征基因,生成特征基因子集。然后通过支持向量机分类模型对特征基因子集进行机器学习训练分类。实验结果表明:急性白血病、结肠癌只需4个特征基因,均获得100%的10折交叉验证分类准确率。最后为了有效地排除噪声基因进而挑选出精确度更高的分类特征基因,采用多尺度小波阈值法对肺癌数据进行降噪处理,降噪后仅需5个特征基因获得96.61%的分类准确率。
关键词:基因表达谱;肿瘤分类;特征基因;信噪比;支持向量机
DNA微阵列技术的出现和发展使肿瘤在分子水平上的研究获得巨大飞跃。研究肿瘤基因表达谱,选取特征基因是从生物信息学角度出发以寻找肿瘤特异基因,在分子水平上准确利用基因表达图谱进行肿瘤亚型识别,对肿瘤的早期诊断和治疗具有重要的实际意义[1-3]。如何对肿瘤基因表达谱进行有效分析、挖掘和发现蕴含的重要信息,已成为近年来生物信息学与系统生物学研究的热点。
自1999年Golub等[4]首次在白血病基因表达谱数据上进行基因识别和肿瘤分类以来,研究者提出多种肿瘤数据挖掘方法。主要有:聚类分析[5]、主成分分析 (PCA)[6]、独立分量分析 (ICA)[7]、k-近邻 (k-NN)[8]、非负矩阵分解 (NMF)[9]、自组织映射 (SOM)[10]、支持向量机 (SVM)[11-13]、人工神经网络 (ANN)[14]、概率神经网络 (PNN)[3]、贝叶斯[15]等经典常用的分类方法。实验表明:分类器的性能对于肿瘤的分类结果至关重要。近年来,基于高效的机器学习方法SVM是该领域最常使用的分类器,对于超高维、小样本特点的肿瘤基因表达谱数据集具有良好的分类效果[16-18]。
本文从肿瘤基因表达谱数据中挖掘有效信息作为分类依据,研究特征基因的选取问题。采用信噪比方法对急性白血病、结肠癌、肺癌基因表达谱数据提取特征基因,利用SVM对3类肿瘤基因样本数据进行训练建立肿瘤分类模型。实验表明:只需4个特征基因急性白血病、结肠癌均获得100%的10折交叉验证分类准确率。最后采用多尺度小波阈值法对肺癌基因表达谱数据进行降噪处理,降噪后只需5个特征基因以96.61%精确度识别肺癌。
1材料与方法
1.1实验数据
使用3类肿瘤数据集:Leukemia、Colon Cancer及Lung Cancer。数据来自哈佛-麻省理工的博德研究所网站http://www.broadinstitute.org/cgi-bin/cancer/datasets.cgi及普林斯顿大学网站http://genomics-pubs.princeton.edu/oncology/。
Leukemia数据集由Golub等收集[4]。它含有72个急性白血病个样本,每个样本含7 129个基因。其中,47个样本为ALL (急性淋巴白血病),25个为AML (急性髓细胞白血病)。选取38个样本作为训练集 (27个ALL,11个AML),34个样本作为测试集 (20个ALL,14个AML)。
Colon Cancer数据集由Alon等收集[19]。它含有62个样本,每个样本含2 000个基因。其中,40个样本为结肠癌,22个为正常组织。随机选取48个样本为训练集 (30个结肠癌,18个正常组织),14个样本为测试集 (10个结肠癌,4个正常组织)。
Lung Cancer数据集由Beer等收集[20]。它含有86个样本,每个样本含7 129个基因。其中,24个样本为肺癌,62个为正常组织。随机选取58个样本为训练集 (16个肺癌,42个正常组织),28个样本为测试集 (8个肺癌,20个正常组织)。
1.2方法
1.2.1信噪比方法
对于肿瘤样本两类别分类问题,信噪比是有效的特征选择方法[4]。公式如下:
其中,u+(j)和u-(j)分别是+1类和-1类样本第j个基因的平均值。类似的,σ+(j)和σ-(j)分别是+1类和-1类样本第j个基因的标准差。
1.2.2支持向量机
SVM是由Vapnik等[21]提出的一种新机器学习方法,它以统计学习理论为基础,基于结构风险最小化原则,在数据小样本条件下具有较好的推广能力。近年来, SVM方法已经成功运用到肿瘤基因表达谱样本数据的挖掘分析之中[12-13]。
支持向量机的具体求解过程如下:
1) 设已知样本训练集:
其中:xi∈X=Rn;yi∈Y={-1,+1}(i=1,2,…,n);xi为特征向量。
2) 选取适当的核函数K(xi,xj),参数C,求解优化问题:
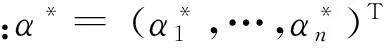
3) 选取α*的正分量,计算样本分类阈值:
4) 构造最优判别函数:
实验采用了Chang等[22]开发的软件包LIBSVM,该软件可通过网址http://www.csie.ntu.etu.tw/~cjlin/libsvm/下载。由于肿瘤样本集非线性的特点,采用基于RBF的SVM分类器对肿瘤样本进行分类。RBF核函数形式为K(xi,xj)=exp(-γ‖xi-xj‖2)。仿真实验环境:Intel(R) Core(TM) i7-4510 CPU @ 2.00GHZ 2.60GHZ 8.00GB的内存,MATLAB R2014a编程实现。
2结果与讨论
本文首先采用信噪比方法对3类肿瘤微阵列数据提取特征基因,然后对提取出的特征基因子集进行归一化,最后以径向基支持向量机作为分类器,利用训练集进行K-折交叉验证,对样本测试集进行基因表达谱数据识别,得到3类肿瘤样本Leukemia、Colon Tumor及Lung Tumor的分类精度。经过多次数值实验,发现白血病数据集提取特征基因最少时仅需要4个 (如表1所示),结肠癌基因表达谱数据集提取征基因最少时仅需要4个 (如表2所示),肺癌数据集提取征基因最少时仅需要5个 (如表3所示),3类肿瘤样本能获得较高的分类精度。

表1 急性白血病特征基因及其生物属性描述
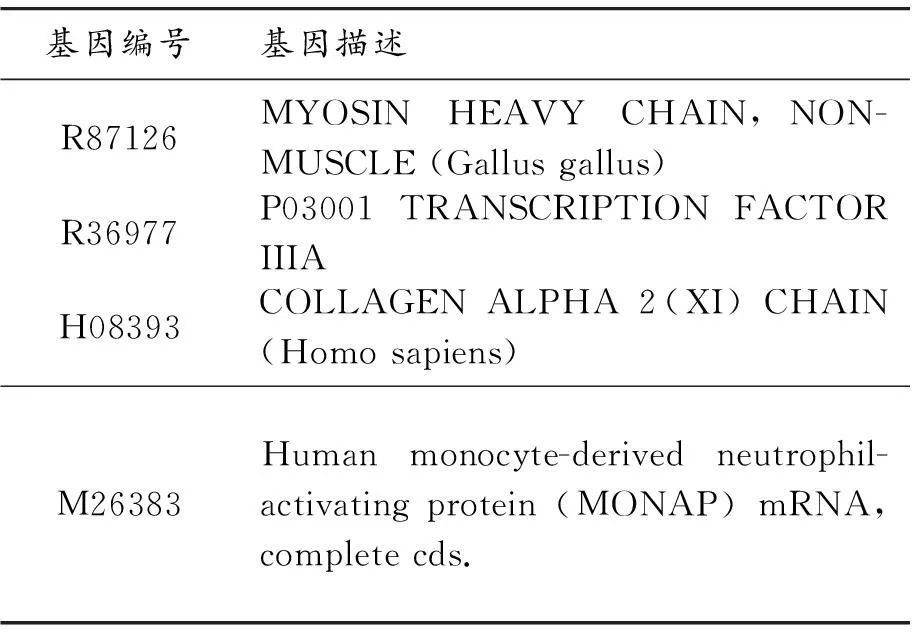
表2 结肠癌特征基因及其生物属性描述

表3 肺癌特征基因及其生物属性描述
在参数选取方面,利用基于SVM肿瘤基因表达谱数据分类模型,结合K-折交叉验证方法,使用LIBSVM软件包中的SVMcgForClass函数对参数进行自动化最优选取。在不同折数选取的最优参数下,急性白血病、结肠癌、肺癌3种癌症的测试集分类准确率如表4所示。

表4 3种癌症分类准确率 %
从表4可以看到:使用不同折数选取的最优参数,对提高癌症分类的准确率具有较大影响。急性白血病的分类在4,5,7,8,10折下均达到100%;结肠癌的分类在4,5,7,8,10折下均达到100%;肺癌的分类精度在3,5折下达到较高的分类精度83.05%。这说明本文提出的方法在K-折交叉验证下,参数自动化最优选取对提高肿瘤的分类准确率具有较大的帮助。
针对肺癌数据集的分类精度不太理想,猜测可能由于基因表达谱中存在噪声,有的噪声强度甚至较大,对含有噪声的基因表达谱提取特征基因时会产生偏差。本研究使用多尺度小波阈值法进行降噪,采用了常用的Daubechies (dbN) 小波系作为母小波,并且在5个不同尺度水平下进行小波重构。分别采用penalty阈值、Birge-Massart阈值、缺省阈值对样本数据进行降噪。通过对肺癌训练集中数据的分析,结果表明:penalty阈值函数的均方根最大,误差最小,分解层数为4时消噪效果最好,采用db4为最佳小波基。将降噪后的微阵列数据采用信噪比方法提取特征基因,最终得到最少时5个特征基因,此时肺癌测试集在3,4,5,7,8,10折下均达到96.61%的分类精确度。降噪后提取的特征基因以及其生物属性描述如表5所示。

表5 肺癌降噪后提取的特征基因
通过对3类肿瘤基因表达谱的研究,可以发现3类数据在10折交叉验证的情况下均达到了最优分类效果,以下研究都采用10折交叉验证选取最优参数。3种肿瘤样本在10折交叉验证下的分类准确率及相应参数如表6所示。

表6 3种肿瘤的分类准确率及相应参数
以结肠癌为例,该数据是一个较难分析的数据集,分别提取了1,2,3,…,10个特征基因做分类研究,研究结果如表7所示。根据不同特征基因个数绘制了分类准确率的折线图,如图1所示。从表7和图1中可以看出:当提取4,6,7,8,10个特征基因时,测试集样本的10折交叉验证分类准确率均达到100%。
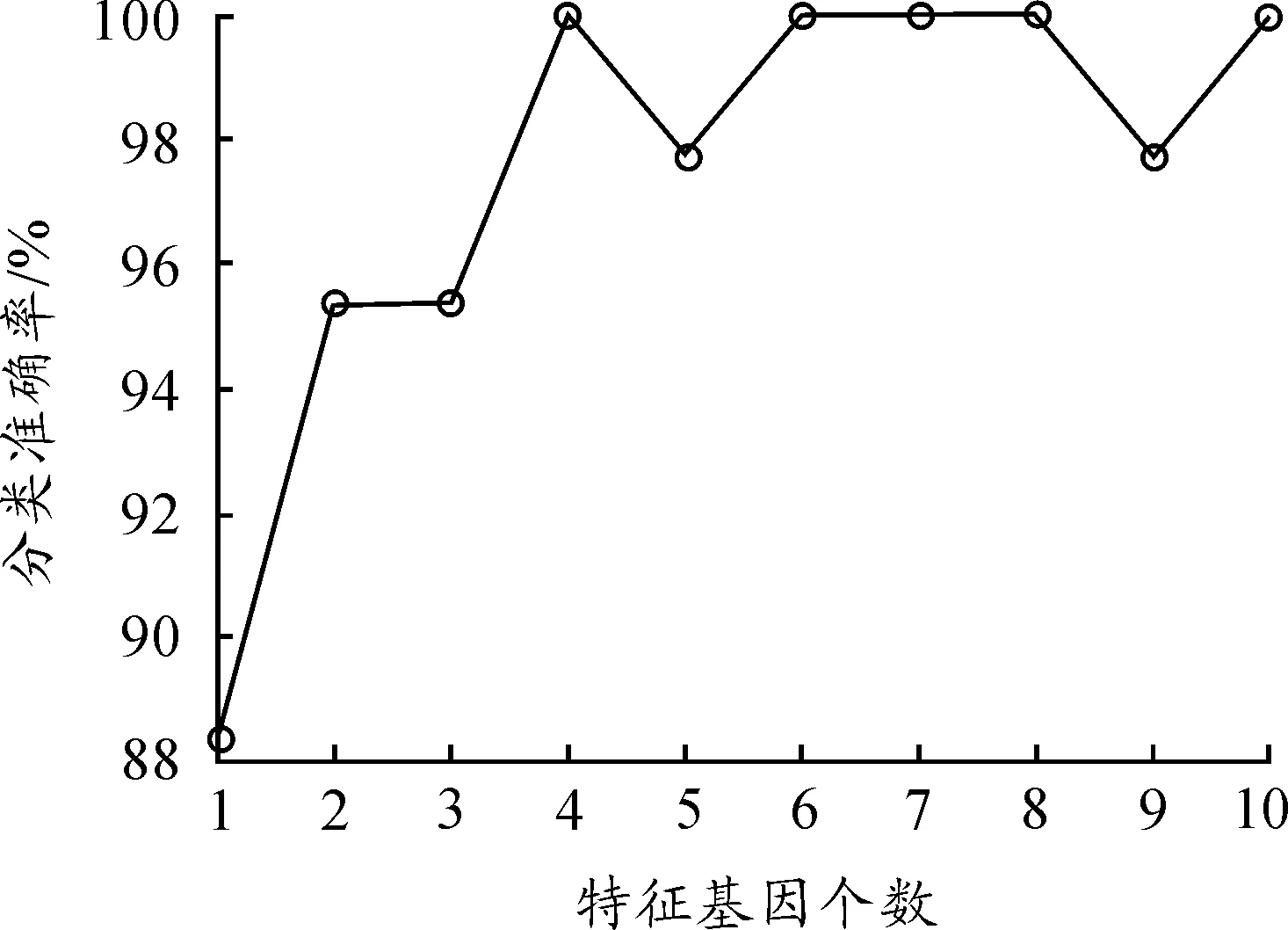
图1 结肠癌的分类准确率
在3种不同肿瘤提取出的特征基因中,对每类肿瘤随机选取3个特征基因绘制三维散点图。图2绘制了急性白血病从特征基因中抽取3基因子集{X95735,M84526,M23197}的三维散点图。图3绘制了结肠癌从特征基因中抽取3基因子集{R87126,R36977,H08393}的三维散点图。图4绘制了肺癌从特征基因中抽取3基因子集{U22816,X04706_s,L43631}的三维散点图。可以看出:图2、3、4中3类肿瘤的样本划分边界都比较清晰,说明该方法提取的特征基因可以将肿瘤较好地区分开。
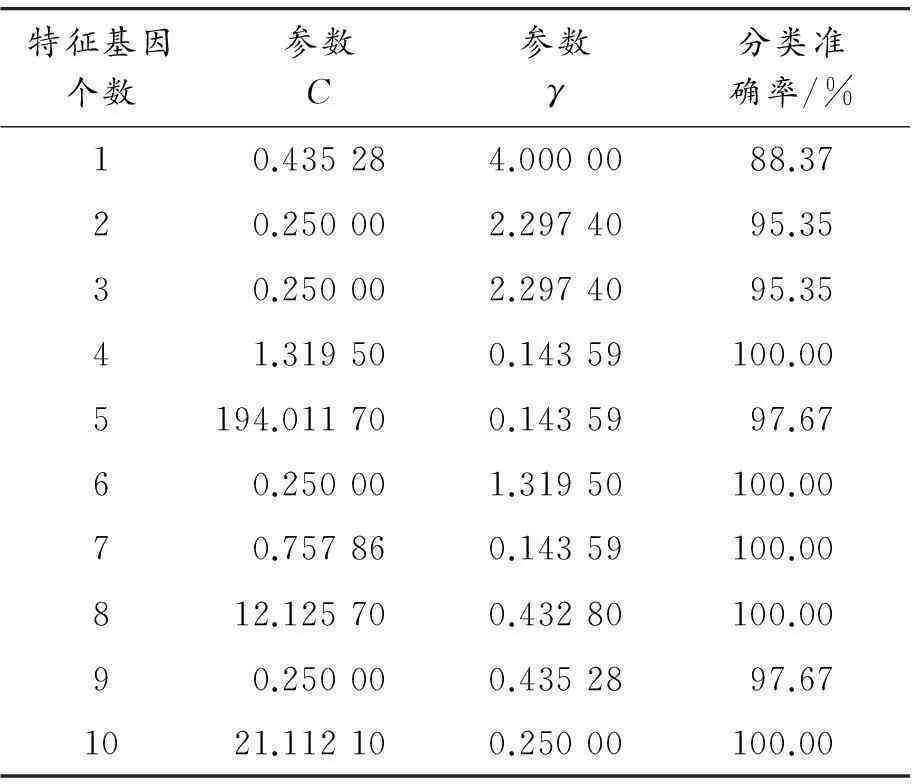
表7 结肠癌选取不同特征基因的分类准确率

图3 结肠癌的3个基因{R87126,R36977,

图4 肺癌数据集的3个基因{U22816,X04706_s,
为便于比较,本文列出了急性白血病、结肠癌、肺癌3类肿瘤样本集采取不同的特征基因提取方法及不同分类器的实验结果,这些都是目前肿瘤分类问题研究中获得的非常好的实验结果,如表8所示。通过比较,可以发现本文提出的方法对两类肿瘤样本的分类准确率均达到较高的水平,相对其他方法实现该方法以最少的特征基因数目达到最高的分类准确率。说明使用RBF核函数的支持向量机作为分类器,采用最优参数选择方法能显著提高样本的分类准确率。

表8 3类肿瘤数据集的不同分类方法获得的分类结果比较
3结束语
肿瘤大数据可以使人们深入了解疾病的病因和结局,为精准医学寻找更好的药物靶点,并且提高疾病的早期预测和预防能力。本文提出一种基于支持向量机的肿瘤基因样本分类模型,针对3类肿瘤基因样本数据集具有样本小、维数高、非线性等特点,利用信噪比和小波降噪等方法对基因表达谱进行降维,提取特征基因子集,运用基于RBF核函数的支持向量机作为分类器。实验结果表明:3类肿瘤样本均获得了较高的分类精确度。不过本文提出的方法适于肿瘤基因表达样本数据的两类别分类,如何利用基于统计学习理论的支持向量机及贝叶斯统计方法建立肿瘤多类别分类模型,并且融入临床生物学信息是下一步的研究方向。
参考文献:
[1]NGUYEN D V,ROCKE D M.Tumor classification by partial least squares using microarray expression data [J].Bioinformatics,2002,18(1):39-50.
[2]YU B,ZHANG Y.The analysis of colon cancer gene expression profiles and the extraction of informative genes [J].J Comput Theor Nanosci,2013,10(5):1097-1103.
[3]WANG S L,LI X L,ZHANG S W,et al.Tumor classification by combining PNN classifier ensemble with neighborhood rough set based gene reduction [J].Computers in Biology and Medicine,2010,40:179-189.
[4]GOLUB T R,SLONIM D K,TAMAYO P,et al.Molecular classification of cancer:class discovery and class prediction by gene expression monitoring [J].Science,1999,5439 (286):531-537.
[5]ZHANG H P,YU C Y,SINGER B,et al.Recursive partitioning for tumor classification with gene expression microarray data [J].Proc.Natl Acad.Sci.,USA,2001,98:6730-6735.
[6]PINTO DA COSTA J F,ALONSO H,ROQUE L.A weighted principal component analysis and its application to gene expression data [J].IEEE/ACM Transactions on Computational Biology and Bioinformatics,2011,8(1):246-252.
[7]HUANG D S,ZHENG C H.Independent component analysis based penalized discriminant method for tumor classification using gene expression data [J].Bioinformatics,2006,22(15):1855-1862.
[8]CHENG X P,CAI H M,ZHANG Y,et al.Optimal combination of feature selection and classification via local hyperplane based learning strategy [J].BMC Bioinformatics,2015,16:219.
[9]ZHENG C H,HUANG D S,ZHANG L,et al.Tumor clustering using non-negative matrix factorizatiowith gene selection [J].IEEE Transactionson Information Technology in Biomedicine,2009,13(4):599-607.
[10]TORONEN P,KOLEHMAINEN M,WONG G,et al.Analysis of gene expression data using self-organizing maps [J].FEBS Letter,1999,451:142-146.
[11]FUREY T S,CRISTIANINI N,DUFFY N,et al.Support vector machine classification and validation of cancer tissue samples using microarray expression data [J].Bioinformatics,2000,16(10):906-914.
[12]PENG S,XU Q,LING X B,et al.Molecular classification of cancer types from microarray data using the combination of genetic algorithms and support vector machines [J].FEBS Letter,2003,555(2):358-362.
[13]YU B,ZHANG Y,ZHAO L K.Cancer classification by a hybrid method using microarray gene expression data [J].J.Comput.Theor.Nanosci.,2015,12(10):3194-3200.
[14]KHAN J,WEI J S,RINGNER M,et al.Classification and diagnostic prediction of cancers using gene expression profiling and artificial neural networks [J].Nature Medicine,2001,7:673-679.
[15]WANG Y,MAKEDON F,FORD J C,et al.Hykgene:a hybrid approach for selecting marker genes for phenotype classification using microarray gene expression data [J].Bioinformatics,2005,21(8):1530-1537.
[16]YU B,LI S,LIU H J.A hybrid gene selection method for tumor classification based on genetic algorithm and support vector machine [J].J.Comput.Theor.Nanosci.,2015,12(11):4730-4735.
[17]SUN S,PENG Q,SHAKOOR A.A kernel-based multivariate feature selection method for microarray data classification [J].PLoS ONE,2014,9(7):e102541.
[18]SHI P,RAY S,ZHU Q F,et al.Top scoring pairs for feature selection in machine learning and applications to cancer outcome prediction [J].BMC Bioinformatics,2011,12:375.
[19]ALON U,BARKAI N,NOTTERMAN D A,et al.Broad patterns of gene expression revealed by clustering analysis of tumor and normal colon tissues probed by oligonucleotide arrays [J].Proc.Natl.Acad.Sci.USA,1999,96:6745-6750.
[20]BEER D G,KARDIA S L R,HUANG C C,et al.Gene expression profile predicts survival of patients with lung adenocarcinoma [J].Nature Medicine,2002,8:816-824.
[21]VAPNIK V N.The nature of statistical learning theory [M].New York:Springer-Verlag New York Inc,1995.
[22]CHANG C C,LIN C J.LIBSVM:a library for support vector machines [J].ACM Transactions on Intelligent Systems and Technology,2011; 2(3):1-27.
[23]ANTONIADIS A,LAMBERT-LACROIX S,LEBLANC F.Effective dimension reduction methods for tumor classification using gene expression data [J].Bioinformatics,2003,19(5):563-570.
(责任编辑何杰玲)
Extraction of Cancer Informative Genes and Gene Expression Data Analysis Based on Support Vector Machine
TAN Yun1, YU Bin2, WANG Qi-ran2, WANG Xue-min2, LI Shan2, QIU Wen-ying2
(1.School of Biological & Chemical Engineering, Qingdao Technical College,Qingdao 266555, China; 2.College of Mathematics & Physics,Qingdao University of Science & Technology, Qingdao 266061, China)
Abstract:This paper put forward cancer gene expression profile data mining methods based on support vector machine (SVM). Firstly, informative genes were extracted from leukemia, colon cancer and lung cancer data by signal-to-noise ratio method, thus generating informative genes subsets. Then informative genes subsets were classified by machine learning and training through support vector machine (SVM) classification model. The experimental results show that only four informative genes are needed for acute leukemia and colon cancer to get 100% classification accuracy by 10 fold cross-validation. Finally, multi-scale wavelet threshold denoising method was established to reduce the noise of the data in lung cancer gene expression profiles for getting higher classification accuracy. After noise reduction, only five informative genes are needed to get 96.61% classification accuracy.
Key words:gene expression profile; cancer classification; informative gene; signal to noise ratio; support vector machine
收稿日期:2016-02-24
基金项目:国家自然科学基金资助项目(41204115); 山东省自然科学基金资助项目(ZR2013AM007, ZR2014FL021); 山东省高等学校科技计划项目(J13LI54)
作者简介:谭云(1979—),女,讲师,主要从事生物信息学的研究;通讯作者 于彬(1976—),男,副教授,硕士生导师,主要从事生物信息学、系统生物学及计算智能的研究。
doi:10.3969/j.issn.1674-8425(z).2016.06.017
中图分类号:Q811.4
文献标识码:A
文章编号:1674-8425(2016)06-0102-07
引用格式:谭云,于彬,王琦然,等.基于SVM的肿瘤特征基因提取与基因表达数据分析[J].重庆理工大学学报(自然科学),2016(6):102-108.
Citation format:TAN Yun, YU Bin, WANG Qi-ran,et al.Extraction of Cancer Informative Genes and Gene Expression Data Analysis Based on Support Vector Machine[J].Journal of Chongqing University of Technology(Natural Science),2016(6):102-108.
猜你喜欢
现代仪器与医疗(2022年1期)2022-04-19
数学物理学报(2020年3期)2020-07-27
北京航空航天大学学报(2019年9期)2019-10-26
雷达学报(2017年3期)2018-01-19
中国水运(2016年11期)2017-01-04
软件导刊(2016年11期)2016-12-22
电子技术与软件工程(2016年20期)2016-12-21
价值工程(2016年32期)2016-12-20
价值工程(2016年29期)2016-11-14
科学与财富(2016年28期)2016-10-14
