
发作性运动诱发性运动障碍一家系临床与遗传学特点
2016-08-03 07:29毕光辉曲星华张慧芳孙淑贞
中国神经精神疾病杂志 2016年4期
毕光辉 曲星华张慧芳 孙淑贞
发作性运动诱发性运动障碍一家系临床与遗传学特点
毕光辉*曲星华*张慧芳*孙淑贞*
目的探讨家族性发作性运动诱发性运动障碍(paroxysmal kinesigenic dyskinesia,PKD)临床及遗传学特点。方法 对1个PKD家系共14名成员进行PRRT2基因检测及调查随访,其中患病2例(1例住院治疗,另1例未治疗),总结分析其临床表现、遗传特点、药物治疗效果及预后。结果该家系2例患者均为男性,患病率14.3%,其中1例不治自愈,1例用卡马西平疗效显著,用拉莫三嗪也有效。该家系为单纯性PKD家系,PRRT2基因检测结果显示该家系中3例存在突变c.797G>A(p.266R>Q),其中1例无临床症状,符合常染色体显性遗传,伴不全外显,存在遗传早现;该家系合并存在多囊肾家族史。结论单纯家族性PKD抗癫痫药物疗效与突变类型及临床特征有关;治疗方案选择应以临床特点及突变类型为依据。
运动诱发性运动障碍 临床特征 遗传学 抗癫痫药
【Abstract】Objective To study the clinical and genetic features of familial paroxysmal kinesigenic dyskinesia (PKD).Methods The clinical information of 14 family members in one pedigree,including 2 patients(one treated in hos⁃pital,the other not treated)were analyzed and the response to treatment and prediction were followed up.DNA was ex⁃tracted from peripheral blood samples,and then screened for PRRT2 mutations.Results There were two male patients in the pure PKD pedigree,Prevalence rate was 14.3%,One of the PKD patients showed good response to carbamazepine as well as lamotrigine whereas other patients recovered without treatment.We detected a nonsense mutation c.797G>A (p.266R>Q)in PRRT2 gene in three family members.One affected member harboring PRRT2 mutation resulted from the incomplete penetrance of the disease,PKD and polycystic kidney disease coexist in the pedigree which showed autoso⁃mal dominant inheritance with incomplete penetrance and anticipation.Conclusions The curative effect of antiepileptic drugs to purely familial PKD is related to mutations and clinical features;Treatments should be decided based upon clini⁃cal features and mutations.
【Key words】Paroxysmal kinesigenic dyskinesias Clinical features Hereditary Antiepileptic drugs
发作性运动障碍(paroxysmal dyskinesias,PxDs)是一组少见的神经系统发作性疾病,目前根据诱发因素可将其分为4种类型,即发作性运动诱发性运动障碍(paroxysmal kinesigenic dyskinesia,PKD)、发作性过度运动诱发性运动障碍、发作性非运动诱发性运动障碍及发作性夜间睡眠性运动障碍[1-3]。PKD是最常见的发作类型,是一种高度异质性疾病,该病的首个致病基因PRRT2(proline⁃rich transmembrane protein 2)于2011年被克隆[4-6]。本文对一单纯PKD家系进行研究,旨在进一步了解其临床及其遗传学特点。

表1引物序列及反应条件
1 资料与方法
1.1临床资料 病例1:先证者Ⅲ1现年18岁,中学生,学习成绩优良,13岁起病,因发作性右侧肢体强直1年入院,站立或起跳时发作,课堂被提问或冥想时亦有发作,持续时间30 s左右,发作时及发作间期均无意识障碍,患者发作前有肢体僵硬感,常采取下蹲或左手握右手以终止发作,发作频率约1~30次/d,发作期和间歇期动态脑电图检查结果均正常,血糖及电解质正常。患者既往顺产,无脑外伤史,无婴儿惊厥、良性家族性婴儿惊厥、婴儿惊厥伴阵发性舞蹈手足徐动症个人史,不合并偏头痛、发作性共济失调等发作性疾病,神经系统查体正常,颅脑MRI、头颈部MRA,脑电图、肌电图以及体感诱发电位检查正常,血常规及血生化及甲功系列检查正常,卡马西平疗效显著,拉莫三嗪也有效。符合BRUNO等[2]提出的PKD临床诊断标准。
病例2:患者Ⅱ,现年43岁,非近亲结婚,发病年龄17岁,发作性右侧肢体强直,发作前无先兆,突然起立时发生,紧张及激动时可诱发发作,每日发作1~20次,持续时间10 s左右,发作时及发作间期均无意识障碍,不合并婴儿惊厥、良性家族性婴儿惊厥、婴儿惊厥伴阵发性舞蹈手足徐动症,无偏头痛、无发作性共济失调等发作性疾病;未住院,未行任何治疗,2年后自愈,至今未发病,神经系统查体正常,门诊颅脑MRI、头颈部MRA、脑电图、肌电图以及体感诱发电位检查正常,符合BRUNO等[2]提出的PKD临床诊断标准。目前因尿毒症在我院例行透析治疗。
家族史:本家系有多囊肾家族史,患者Ⅱ及其生母、姨母以及姨表姐均为多囊肾患者。
1.2方法
1.2.1外周血基因组DNA提取 在患者知情同意的基础上,依照有关法律规定并经我院伦理委员会批准,除成员Ⅲ2拒绝外抽取该家系中所有患者及健康家属的外周血各6 mL,采用标准酚-氯仿法提取基因组DNA,TE缓冲液溶解,-20℃保存备用。
1.2.2引物合成 利用Primer 3在线设计软件进行引物设计,涵盖PRRT2(NM_145239)所有外显子编码区及内含子-外显子剪切区。
1.2.3聚合酶链式反应及Sanger测序、比对 采用标准PCR反应体系,于相应的退火温度条件下进行PCR反应,2%琼脂糖凝胶电泳进行反应产物的鉴定及纯化,ABI3730自动测序仪进行PRRT2测序。测序结果应用DNAStar软件与标准序列进行比对分析。

图1 PKD一家系谱图
2 结果
2.1临床特点本研究家系临床特点:①由站立,起跳或紧张、激动、冥想等诱发;②首发症状为右侧肢体强直;③发作时间短10~30 s,发作频率1~30次/d;④发作过程无意识障碍;⑤神经系统查体正常;⑥脑电图、颅脑MRI、MRA及其他辅助检查结果正常;⑦发病年龄13~17岁;⑧对卡马西平疗效显著,拉莫三嗪也有效。符合BRUNO等[2]提出的PKD临床诊断标准:有明确诱发因素,大多由突然某一动作或运动诱发,部分由紧张、惊吓或其他因素诱发;临床特点为肌张力障碍,有的表现为手足徐动、舞蹈样动作,或偏侧投掷等,可单一发作,也可几种发作形式的组合;发作持续时间短暂,一般不超过1 min;发病年龄1~20岁,有家族史的患者可酌放宽发病年龄;抗癫痫药治疗有效,特别是卡马西平及苯妥英钠疗效明显;神经电生理学检查正常,排除其他器质性疾病。
2.2遗传特征 该家系连续2代发病,成员Ⅰ经基因检测为携带者,无症状,成员Ⅱ、Ⅲ1有临床症状,符合常染色体显性遗传伴不全外显,外显率66.7%,先证者Ⅲ1与患者Ⅱ比较,发病年龄提前,病程延长,病情加重,呈遗传早现现象。
2.3基因检测结果 该家系成员Ⅰ、Ⅱ、Ⅲ1的PRRT2基因2号外显子均存在突变c.797G>A (p.266R>Q),突变类型为错义突变,其余成员均未发现基因突变(见图2)。

图2基因测序图
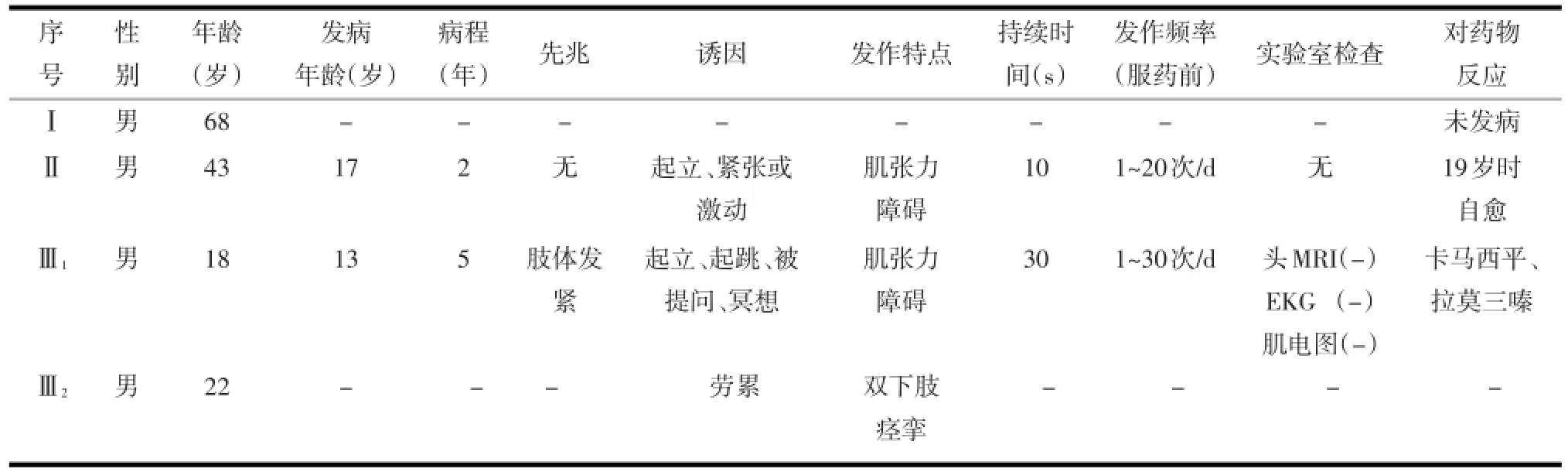
表2 1PKD家系临床特点
3 讨论
PKD是一种少见的运动障碍性疾病,由KERTESZ[3]于1967年首先报告。2004年BRUNO等[2]提出其临床诊断标准;在已报道的PKD病例中,大部分呈家族性,家族性患者的遗传方式以常染色体显性遗传为主,可伴不完全外显,也有呈常染色体隐性遗传的家系报道[7-8]。周瑾瑕等[9-10]曾报道8个PKD家系,其中6个家系为常染色体显性遗传,其中1个家系为不全外显。其他国内报道的一系列PKD家系,也以单纯家系居多,本文报道家系的遗传方式为常染色体显性遗传伴不全外显,不伴发其它发作性疾病,为1单纯PKD家系,而国外家族性发作性动作诱发性运动障碍患者中,单纯发作性动作诱发性运动障碍较为少见,大多伴发其它疾病,如发作性共济失调、婴儿惊厥、婴儿惊厥伴阵发性舞蹈手足徐动症、良性家族性婴儿惊厥、偏头痛等。出现这种差异是否与人种有关有待进一步研究。迄今为止,共有3个位点与发作性动作诱发性运动障碍有关,即EKD1~3[8,11-14]。EKD1定位于 16p11.2~q12.1[13];EKD2定位于 16q13 ~q22.1[8,14];SPACEY等[15]在2002年对一个英国单纯PKD家系进行了研究,发现其位点与EKD1、EKD2不连锁,在16号染色体上未发现相关连锁区域,因此确定有第3个位点存在,命名为EKD3。并认为第3个位点EKD3与单纯性PKD相关[15-16]。2011年,上海瑞金医院运动障碍性疾病课题组及其他来自中国的研究小组率先证实了PRRT2基因为家族性PKD的致病基因[4-6]。本文报道的家系,基因检测结果PRRT2基因2号外显子均存在突变c.797G>A(p.266R>Q),突变类型为错义突变,再次证实PRRT2为家族性PKD致病基因,通过比较发现,本文报道的PKD家系与SPACEY等[15]报道的英国家系发病年龄及临床表现都非常相似,致病基因位点不同,提示存在遗传异质性。
系列研究发现PRRT2基因突变不仅存在于PKD,也存在于其他一系列发作性疾病,如家族性婴儿惊厥伴阵发性舞蹈手足徐动症、热性惊厥、发作性共济失调等[17-18],推测PKD与上述一系列发作性疾病在遗传学方面可能属同一疾病,只是临床表型不同,并由此提出了“PRRT2相关疾病(PRD)”的概念[19-20]。PRRT2基因包含4个外子,其中第2和3外显子为主要突变部位,我们研究的PKD家系基因突变都位于PRRT2基因第2外显子。在PRRT2相关疾病中发现的基因突变类型有56种,目前更有文献报道增至71种,其中主要突变类型为无义突变,其他突变类型包括错义突变、剪切突变和插入突变[11-12]。目前有研究提出,PRRT2基因突变中,c.649dupC(p.R217PfsX8)为热点突变[21],因此,对于PKD家系患者,进行PRRT2基因检测时,c.649dupC(p.R217PfsX8)可列为首选。我们报道的PKD家系致病基因PRRT2,突变c.797G>A(p.266R>Q),突变类型为错义突变,也属较常见的突变类型。FRIEDMAN等[31]及CAI等[32]研究表明携带PRRT2错义突变的PKD患者症状相对较轻微且外显率低;这一点与本研究有所不同,我们报道的PKD家系外显率66.7%,临床症状明显,先证者症状更为突出。LI等[33]对81例PKD患者研究发现,携带PRRT2基因突变与不携带PRRT2基因突变临床表现有很大不同,携带基因突变的PKD患者临床上表现为舞蹈手足徐动症,而不携带基因突变的患者表现为舞蹈手足徐动症或肌张力障碍,这也与我们的研究差异较大;本PKD家系2例患者临床表现均为典型的肌张力障碍,而且诱发因素基本相同。携带基因突变的患者发病年龄更早,每次发作持续时间更长,对药物治疗反应差别较大[33],另外,携带PRRT2基因纯合突变的患者临床表型更严重,伴有失神发作、周期性共济失调及智力低下等。一种突变可导致多种表型,c.649dupC可导致几乎所有的PKD表型[19-20]。因此PRRT2基因突变的基因型与表型之间的关系目前还无法确定,是突变导致的甲基化形式不同或引起修饰方式有差异,待进一步研究。
PKD治疗首选卡马西平,突变携带者对小剂量卡马西平反应完全[33],这与本研究结果基本一致;约94%非基因突变携带者对相同剂量卡马西平反应不完全[33]。本家系患者Ⅲ1对小剂量卡马西平疗效显著,后因年龄较小改为拉莫三嗪,疗效明显弱于卡马西平,这与国内郑乃智等[34]报道有所不同,除了基因突变因素外,首发症状及临床特点决定了疗效的差异性,与PKD家族性或散发是否也有关,有待进一步研究,提示PKD治疗在药物选择上要有针对性。
目前PKD的发病机制仍未明确,有报道PKD患者双侧尾状核存在异常灌注,静息态功能磁共振扫描发现双侧壳核低频振荡,振幅与正常对照相比明显增加,提示PKD发作可能与壳核的异常活动有关;因条件限制本研究未行这两方面检查。在大脑皮质以及小脑、小脑脚、苍白球、尾状核、丘脑底核等结构中可检测到高表达的PRRT2 mRNA[3]。无义突变是PRRT2基因突变主要类型,引起肽链截短,介导mRNA降解,导致PRRT2蛋白功能丧失[23],引起细胞膜定位功能异常,引发PKD[4]。系列研究发现,PRRT2蛋白与突触相关蛋白25(SNAP25)是两种相互作用蛋白[25]。SNAP25参与神经突触囊泡胞吐过程[26]。在电压门控性钙离子通道(VGCC)的调控以及突触前膜钙离子介导的胞吐过程中发挥重要作用。PRRT2蛋白发生功能缺失后,SNAP25功能受到影响,导致电压门控性钙离子通道调控失常,影响突触囊泡胞吐过程诱发疾病的发生。因此推测突触功能异常及神经递质释放可能为PKD的发病机制。
我们报道的PKD家系还存在遗传早现现象。表现为发病年龄提前,先证者Ⅲ1较患者Ⅱ发病年龄提前4岁,且病程延长,病情加重。周瑾瑕等在2006年报道的6个家系中,有4个家系存在遗传早现,其中3个家系为完全外显,1个家系不完全外显[9]。遗传早现临床特点为随代数延续,逐代发病年龄前移,且伴有病情随代加重,病程延长等,孟德尔遗传定律无法解释这一现象,致病基因内三核苷酸重复序列拷贝数不稳定异常扩展可能与遗传早现有关[28],主要表现在显性神经遗传病,如脊髓小脑型共济失调(SCA)、亨廷顿病、面肩肱型肌营养不良等,在PKD遗传早现现象报道较少,可能与PKD发病率低有关,单纯PKD家系发病率更低,再者PKD具临床及遗传异质性合并症多。此外我们报道的PKD家系,成员Ⅰ为基因携带者,成员Ⅱ既是PKD患者,同时又是多囊肾患者,且有多囊肾家族史,调查发现患者Ⅱ生母及姨母以及姨表姐均为多囊肾患者,目前患者Ⅱ因尿毒症在本院例行透析治疗。研究表明多囊肾也是常染色体显性遗传性疾病,具有明显家族史,研究证实85%的患者致病基因在16号染色体[29,31],这一点与PKD有共同之处,因此患者Ⅱ出现PKD临床症状是否与多囊肾也有关系,或者多囊肾促发了PKD症状,另外二者之间在致病基因方面有无关联性,有待于进一步探讨。
致谢:上海交通大学瑞金医院神经内科曹立教授、黄啸君博士、刘晓黎博士、陈生弟教授给予的指导和帮助深表感谢。
[1]DEMIRKIRAN M,JANKOVIC J.Paroxysmal dyskinesias;clini⁃cal features and classification[J].AnnNeurol,1995,38(14):571-579
[2]BRUNO MK,HALLETT M,GWINN-HARDY K,et al.Clinical evaluation of idiopathic paroxysmal kinesigenic dyskinesia:new diagnostic criteria[J].Neurology,2004,63(12):2280-2287.
[3]KERTESZ A.Paroxysmal kinesigenic choreoathetosis.An entity within the paroxysmal choreoathetosis syndrome.Description of 10 cases,including 1 autopsied[J].Neurology,1967,17(7):680-690.
[4]CHEN WJ,LIN Y,XIONG ZQ,et al.Exome sequencing identi⁃fies truncating mutations in PRRT2 that cause paroxysmal kine⁃sigenic dyskinesia[J].Nat Genet,2011,43(12):1252-1255.
[5]LI J,ZHU X,WANG X,et al.Targeted genomic sequencing identifies PRRT2 mutations as a cause of paroxysmal kinesigen⁃ic choreoathetosis[J].J Med Genet,2011,49(2):76-78.
[6]WAN G JL,CAO L,LI XH,et al.Identification of PRRT2 as the causative gene of paroxysmal kinesigenic dyskinesias[J]. Brain,2011,134(Pt 12):3493-3501.
[7]DALE RC,GARDINER A,ANTONY J,et al.Familial PRRT2 mutation with heterogeneous paroxysmal disorders including par⁃oxysmal torticollis and hemiplegic migraine[J].Dev Med Child Neurol,2012,54(10):958-960.
[8]VALENTE EM,SPACEY SD,WALI GM,et al.A second parox⁃ysmal kinesigenic choreoathetosis locus(EKD2)mapping on 16q13-q22.1 indicates a family of genes which give rise to par⁃oxysmal disorders on human chromosome 16[J].Brain,2000,123(Pt 10):2040-2045.
[9]周瑾瑕,李国良,刘鼎,等.家族性发作性运动诱发性运动障碍六个家系的临床及遗传特点分析[J].中华神经科杂志,2006,39(11):726-729.
[10]周瑾瑕,李国良,陈婵娟,等.单纯型家族性发作性运动诱发性运动障碍二家系基因连锁分析[J].中华神经科杂志,2008,41(3):159-163.
[11]LEE HY,HUANG Y,BRUNEAU N,et al.Mutations in the gene PRRT2 cause paroxysmal kinesigenic dyskinesia with in⁃fantile convulsions[J].Cell Rep,2012,1(1):2-12.
[12]LEE YC,LEE MJ,YU HY,et al.PRRT2 mutations in paroxys⁃mal kinesigenic dyskinesia withinfantile convulsions in a Tai⁃wanese cohort[J].PLoS One,2012,7(8):e38543.
[13]TOMITA H,NAGAMITSU S,WAKUI K,et al.Paroxysmal kinesi⁃genic choreoathetosis locus maps to chromosome 16p11.2-q12.1 [J].Am J Hum Genet,1999,65(6):1688-1697.
[14]BENNETT LB,ROACH ES,BOWCOCK AM.A locus for parox⁃ysmal kinesigenic dyskinesia mapsto human chromosome 16 [J].Neurology,2000,54(1):125-130.
[15]SPACEY SD,VALENTE EM,WALI GM,et al.Genetic and clinical heterogeneity in paroxysmal kinesigenic dyskinesia:evi⁃dence for a third EKD gene[J].Mov Disord,2002,17(4):717-725.
[16]ZHOU J,LI G,CHEN C,et al.Familial pure paroxysmal kinesi⁃genic dyskinesia in Han population from the Chinese mainland:a new subtype?[J].Epilepsy Res,2008,80(2/3):171⁃179.
[17]SWOBODA KJ,SOONG BW,MCKENNA C,et al.Paroxysmal kinesigenic dyskinesia and infantile convulsions:clinical and linkage studies[J].Neurology,2000,55(2):224⁃230.
[18]SZEPETOWSKI P,ROCHETTE J,BERQUIN P,et al.Familial infantileconvulsions and paroxysmal choreoathetosis:a new neurological syndrome linked to the pericentromeric region of human chromosome 16[J].Am J Hum Genet,1997,61(4):889-898.
[19]BECKER F,SCHUBERT J,STRIANO P,et al.PRRT2⁃related disorders:further PKD and ICCA cases and review of the litera⁃ture[J].J Neurol,2013,52(12):980-987.
[20]MÉNERET A,GAUDEBOUT C,RIANT F,et al.PRRT2 muta⁃tions and paroxysmal disorders[J].Eur J Neurol,2013,2013,63 (13):1274-281.
[21]CAO L,HUANG XJ,ZHENG L,et al.Identification of a novel PRRT2 mutation in patients with paroxysmal kinesigenic dyski⁃nesias and c.649dupC as a mutation hot⁃spot[J].Parkinsonism Relat Disord,2012,18(5):704⁃706.
[22]NICHOLSON P,YEPISKOPOSYAN H,METZE S,et al.Mühle⁃mann Nonsense⁃mediated mRNA decay in human cells:mecha⁃nistic insights,functions beyond quality control and the double⁃life of NMD factors[J].Cell Mol Life Sci,2010,67(4):677⁃700.
[23]CARTEGNI L,CHEW SL,KRAINER AR.Listening to silence and understanding nonsense:exonic mutations that affect splic⁃ ing[J].Nat Rev Genet,2002,3(3):285⁃298.
[24]LEE HY,NAKAYAMA J,XU Y,et al.Dopamine dysregulation in a mouse model of paroxysmal nonkinesigenic dyskinesia[J].J Clin Invest,2012,122(1):507⁃518.
[25]STELZL U,WORM U,LALOWSKI M,et al.A human protein⁃protein interaction network:a resource for annotating the pro⁃teome[J].Cell,2005,122(5):957⁃968.
[26]RAJAKULENDRAN S,KASKI D,HANNA MG.Neuronal P/Q⁃type calcium channel dysfunction in inherited disorders of the CNS[J].Nat Rev Neurol,2012,8(2):86⁃96.
[27]CORRADINI I,DONZELLI A,ANTONUCCI F,et al.Epilepti⁃form activity and cognitive deficits in SNAP⁃25+/-mice are normalized by antiepileptic drugs[J].Cereb Cortex,2012,10 (12):2076-2082.
[28]MADDOX J.Triplet repeat genes raise questions[J].Nature,1994,368(6):685.
[29]HUGHES J,WARD CJ,PERAL B,et al.The polycyctic kid⁃ney disease 1(PKD1)gene encodes a novel protein with multi⁃plecellrecognitiondomains[J].NatGenet,1995,10(2):151-160.
[30]TORRES,VICENTE,HARRIS,et al.Autosomal dominant polycyctic kidney disease:the last 3 years[J].Kidney Interna⁃tional,2009,76(2):149-168.
[31]Friedman J,Olvera J,Silhavy JL,et al.Mild paroxysmal kinesi⁃genic dyskinesias caused by PRRT2 missense mutations with re⁃duced penetrance[J].Neurology,2012,79(9):946-948.
[32]CAI C,SHI O,LI WD.Missense mutations ofthe proline-rich transmembrane protein 2 gene cosegregate with mild paroxys⁃mal kinesigenic dyskinesia and infantile convulsions in a Chi⁃nese pedigree[J].Parkinsonism Relat Disord,2013,19(3):402-403.
[33]LI HF,CHEN WJ,NI W,et al.PRRT2 mutation correlated with phenotype of paroxysmal kinesigenic dyskinesia and drug response[J].Neurology,2013,80(16):1534-1535.
[34]郑乃智,王敏,宋旭霞,等.拉莫三嗪治疗发作性运动诱发性运动障碍1 4例临床疗效观察[J].临床神经电生理学杂志,2009,18(2):84-86.
(责任编辑:李立)
Analysis of clinical and genetic features of one family with paroxysmal kinesigenic dyskinesia.
BI Guanghui,QU Xinghua,ZHANG Huifang,SUN Shuzhen,Department of Neurology,Dong ying people's Hospital,Dongying 257091,China.Tel:0546-8901185.
R748
A
10.3969/j.issn.1002-0152.2016.04.005
*东营市人民医院(山东东营257091)
(E-mail:BGHU66@163.com)
2015-12-14)
猜你喜欢
广西林业科学(2022年6期)2023-01-16
中华实用诊断与治疗杂志(2022年1期)2022-08-31
中华实用诊断与治疗杂志(2022年1期)2022-08-31
中国神经精神疾病杂志(2022年3期)2022-07-14
世界科学技术-中医药现代化(2021年8期)2021-12-21
浙江科技学院学报(2020年4期)2020-08-01
中国医院用药评价与分析(2018年9期)2018-01-17
中国卫生标准管理(2015年25期)2016-01-14
郑州大学学报(医学版)(2015年2期)2015-02-27
中国卫生标准管理(2015年13期)2015-01-26
