
TRAIL诱导细胞死亡及其耐药机制研究进展
2016-11-09 02:27乔新然陈淑珍
中国医药生物技术 2016年5期
乔新然,陈淑珍
TRAIL诱导细胞死亡及其耐药机制研究进展
乔新然,陈淑珍
肿瘤坏死因子相关的凋亡诱导配体(tumor necrosis factor-related apotosis-inducing ligand,TRAIL/Apo-2L)是继肿瘤坏死因子(tumor necrosis factor,TNF)、Fas 配体(Fas ligand,FASL)之后发现的又一 TNF 超家族成员,广泛分布于机体中,在自身免疫疾病及抗肿瘤中扮演重要角色。TRAIL 因其能够选择性地靶向肿瘤细胞而对正常细胞不产生毒性,成为近年来抗肿瘤研究的热点。但随着临床研究的深入,TRAIL 的单药治疗在多种肿瘤细胞中出现耐药现象,这阻碍了 TRAIL 在抗肿瘤治疗中的应用。因此,本文就TRAIL 诱导细胞死亡机制、细胞耐药机制及联合用药策略进行综述。
1 TRAIL 及其受体
1.1 TRAIL/Apo-2L
TRAIL/Apo-2L 是一个 33 ~ 35 kD 的 II 型跨膜蛋白,由 281 个氨基酸残基组成,胞外羧基端所具有的高度保守TNF 同源结构域(TNF homology domain,THD)与三聚体的形成有关。在生理条件下,TRAIL/Apo-2L 分子以完整的膜结合形式存在于免疫效应细胞(如 T 细胞)的细胞膜上或者以截断的可溶性形式(残基 114 ~ 281)被尿激酶纤溶酶激活物(uridylyl phosphate adenosine,uPA)释放。这两种形式均具有生物活性,都能与死亡受体形成三聚体,并由锌离子稳定,产生凋亡诱导作用。
1.2 TRAIL 受体
TRAIL 受体属于 TNF 受体超家族,分为三种:
⑴死亡受体(death receptor):包括 TRAIL-R1/DR4 和TRAIL-R2/DR5,分别是由 468 个和 411 个氨基酸残基组成的 I 型跨膜蛋白,有 58% 的一致性,在机体内分布广泛。两者的胞外区均有 2 个富含半胱氨酸的假重复序列,胞内区有与 TNFR、Fas 同源的死亡结构域(death domain,DD)。
⑵诱骗受体(decoy receptor):包括 TRAIL-R3/DcR1 和TRAIL-R4/DcR2。DcR1 是由 299 个氨基酸残基组成的I 型跨膜蛋白,其胞外区与 DR4、DR5 类似,存在 2 个富含半胱氨酸的序列,没有跨膜区和胞内区,仅通过一个糖酯磷脂酰肌醇(glycosyl phosphatidyl inositol,GPI)连接在细胞表面。因此,它与 TRAIL 结合后不能诱导凋亡。DcR2是由 386 个氨基酸残基组成的 I 型跨膜蛋白,存在跨膜区和一段被剪切的 DD,也无法传递凋亡信号。
⑶骨保护素(osteoprotegerin,OPG):TRAIL 的第 3 个诱骗受体,由 401 个氨基酸残基组成的胞外区可溶性受体,与 TRAIL 有较弱的结合能力,能够抑制 TRAIL 所诱导的凋亡[1-2]。
2 TRAIL 诱导细胞死亡的机制
2.1 TRAIL 诱导细胞凋亡
TRAIL 与功能性受体 DR4/DR5 结合后,活化形成三聚体,通过 DD 相互作用,募集死亡结构域连接蛋白(Fas associated death domain,FADD)到受体上,再通过 FADD中的死亡效应结构域(death effector domain,DED)募集启始 caspases(pro-caspase-8/10)到受体复合物上,形成死亡诱导信号复合体(death-inducing signaling complex,DISC)。Pro-caspase-8/10 在 DISC 中自我剪切活化,形成活性caspase-8/10 并释放到细胞质中,激活下游 caspase 信号级联[1,3],诱导细胞凋亡。
由于内源性 caspase 抑制剂 X 染色体连锁凋亡抑制蛋白(X-linked inhibitor of apoptosis protein,XIAP)的表达极大地影响 DISC 中 caspase 的活化,因此根据 caspase活化程度及时间的不同,将细胞分为 I 型和 II 型。在 I 型细胞中,caspase-8/10 充分活化,受 XIAP 的抑制较小,能直接激活效应 caspases(caspase-3/6/7),诱导细胞凋亡的外在途径(extrinsic pathway)。在 II 型细胞中,XIAP 的表达只允许少量活性 caspase-8/10 的形成,需要激活线粒体介导的内在凋亡途径(intrinsic pathway):少量活性 caspase-8/10分裂 BH3-only 蛋白(BCL-2 inhibitory BH3-domain containing protein,BID)形成 tBID(truncated BID),tBID 运输到线粒体,激活 BAX(Bcl-2 associated X protein)和 BAK(Bcl-2 antagonist killer 1),进而诱导线粒体释放前凋亡因子(如细胞色素 c、SMAC/DIABLO、HTRA2/Omi)到细胞质中,细胞色素 c、凋亡蛋白活性因子-1(apoptosis activating factor-1,APAF-1)和 pro-caspase-9 在细胞质中组成凋亡小体,能够激活另一个启始 caspase——caspase-9,从而进一步激活效应 caspase-3/7,引起细胞凋亡[1,4]。SMAC/DIABLO的释放能够抑制 XIAP 的活性,进一步促进凋亡(图 1)。
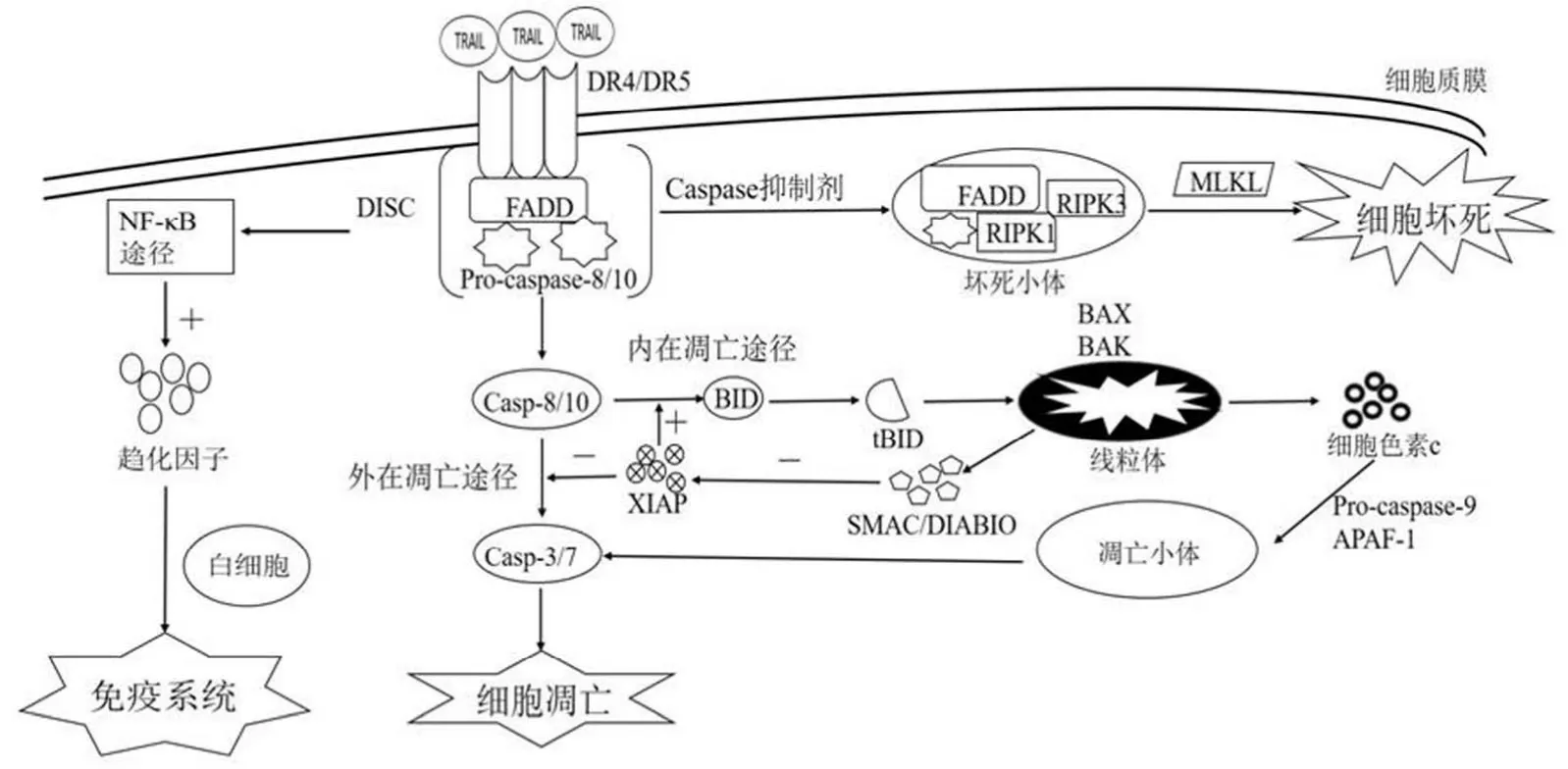
图1 TRAIL 诱导细胞死亡的机制
2.2 TRAIL 与自噬
2.2.1 自噬的双面性 细胞自噬是细胞一种自我降解的过程,在肿瘤细胞中,既是细胞防御机制,又是程序性细胞死亡机制。近年来发现,自噬在 TRAIL 引起的肿瘤耐药中有着重要的作用。
2.2.1.1 抑制自噬——增强 TRAIL 抗肿瘤活性 研究发现,乳腺癌细胞长期暴露在 TRAIL 中,自噬增加,产生耐药性,当 TRAIL 与自噬抑制剂氯喹联合作用于 TRAIL 耐药的乳腺癌细胞时,能扭转这种耐药性,使细胞对 TRAIL敏感,从而增强了抗肿瘤效果[5-6]。烟酸作用于人结肠癌细胞 HCT-116 时,能够激活自噬流,使细胞逃离 TRAIL 引起的线粒体膜电位功能紊乱及细胞死亡。这表明,抑制自噬能增强 TRAIL 的抗肿瘤活性[7]。
2.2.1.2 激活自噬——增强 TRAIL 抗肿瘤活性 激活自噬也能增强 TRAIL 的抗肿瘤活性[8]。例如查耳酮衍生物chalcone-24(Chal-24)通过激活肺癌细胞中的自噬来引起c-FLIPL 和 c-IAPs 的降解,进而增强 TRAIL 的抗肿瘤活性[9]。在甲状腺癌中,增加自噬能使 TRAIL 敏感的 TPC-1细胞对 TRAIL 更加敏感,产生更好的抗肿瘤效果,而对于TRAIL 耐药的 FRO 细胞,抑制自噬才能使其对 TRAIL敏感[10],若封闭 TRAIL 敏感细胞中的自噬作用能够降低对TRAIL 的敏感度,因此,保留基本的自噬水平对 TRAIL 诱导的凋亡是必需的[11]。
2.2.2 自噬与 TRAIL 介导的细胞凋亡之间的机制
2.2.2.1 自噬小体和 p62 蛋白的积累 TRAIL 能够激活TRAIL 耐药细胞中的自噬流,导致 p62 降解并阻止caspase-8 激活,但在 TRAIL 敏感细胞中,TRAIL 能抑制自噬流,从而导致 caspase-8 活化和细胞凋亡[12]。研究发现,在自噬过程中的不同阶段抑制自噬能导致 TRAIL 敏感的前列腺癌 PC3 细胞中对于 TRAIL 诱导的细胞死亡产生不同结果:在 LC3 与自噬体 l 膜联合之前,使用 3-甲基腺嘌呤来抑制自噬,能够阻止 p62 的聚合和 caspase-8 的激活,从而抑制细胞死亡;而使用 CQ 来抑制自噬小体降解,并没有影响 TRAIL 诱导的 PC3 细胞死亡。这表明,在缺乏自噬降解的情况下,自噬小体和 p62 蛋白质聚合物的积累对于 TRAIL 诱导的细胞死亡是必需的,在 TRAIL敏感细胞中自噬降解或许处于被抑制的状态。
2.2.2.2 活性 caspase-8 的自噬溶酶体降解 活性caspase-8 的自噬溶酶体降解被认为是自噬和 TRAIL 诱导细胞凋亡的中间机制[10]。例如 TRAIL 引起 DNA 损坏,使 PARP 被激活,进而发生 HMBG1 PARylation,HMBG1迁移到细胞质中与 BECN-1 结合,启动了自噬,减弱了caspase-8 活性,产生 TRAIL 耐药,抑制 PARP1-HMGB1途径能够减弱自噬,从而增加 TRAIL 引起的凋亡。因此,TRAIL 耐药细胞耐药的原因之一是由于不同途径启动的自噬导致活性 caspase-8 的缺失[13](图 2)。
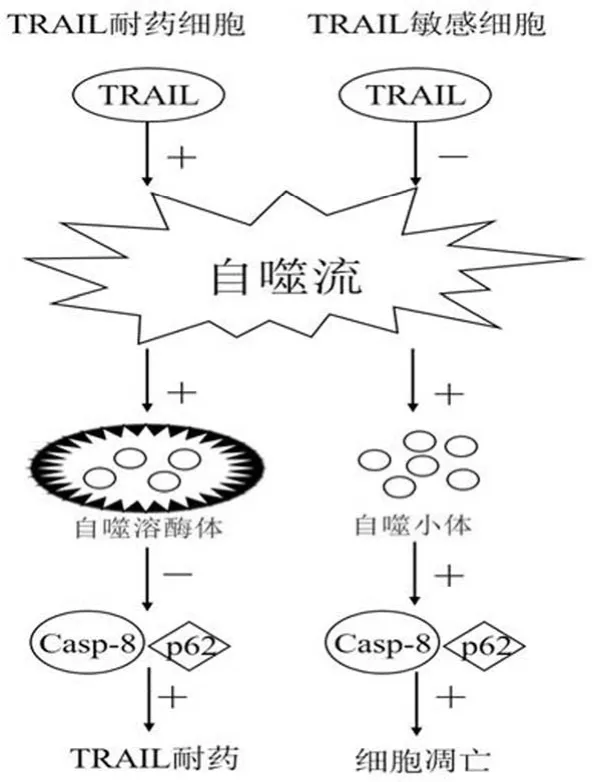
图2 自噬与 TRAIL 介导的细胞凋亡之间的机制
2.2.2.3 Beclin 1 和生存素相互作用 Beclin 1 和生存素之间的相互作用可能是自噬和凋亡之间另一个潜在的机制。实验表明,敲低 Beclin 1 能下调生存素,使神经胶质瘤细胞对 TRAIL 诱导的细胞凋亡敏感,当引入生存素时能抵抗这种敏化效果,这表明下调生存素能增强细胞对 TRAIL 诱导凋亡的敏感性[14]。
2.3 TRAIL 诱导细胞坏死
当用 caspase 抑制剂来抑制 caspase 活性时,TRAIL虽然无法诱导凋亡,但可以诱导受体相互作用蛋白激酶(receptor interacting protein kinase,RIPK)介导的细胞死亡,称为程序性坏死[3],这种类型的细胞死亡依赖于死亡小体的激活。当 caspase-8 缺失或被抑制时,RIPK1 和 RIPK3 通过 RIP 同型结构域(RIP homotypic interaction motif,RHIM)相连接形成死亡小体,RIPK3 在死亡小体中自身磷酸化激活,募集并磷酸化混合谱系激酶(mixed lineage kinase-like,MLKL),使 MLKL 发生构象变化,但是对于MLKL 诱导的凋亡性坏死的分子机制目前仍不完全清楚。有研究表明,MLKL 可能参与破坏和调节细胞膜孔的变形(图 1)。
2.4 TRAIL 调节免疫系统
TRAIL 能够在巨噬细胞、树突细胞、T 细胞、NK 细胞中表达,在自身免疫性疾病、细菌和病毒感染、肿瘤新陈代谢的免疫监视中起重要作用[4]。TRAIL 通过 NF-κB 途径诱导趋化因子 CXCL2、CCL4、CCL20 的分泌,这些趋化因子在生理环境下募集白细胞,从而调节炎症[15](图 1)。TRAIL/TRAIL-R 途径涉及免疫系统的调控与稳定。例如,在大脑中,TRAIL-DR5 信号通路能调控炎症、神经增殖分化以及调节低氧-组织缺血后的脑损伤。TRAIL 受体缺陷的老鼠更容易产生 DSS(dextran sulphate sodium)诱导的结肠炎和结肠炎相关的癌变,并且在结肠炎期间,对免疫细胞有显著的渗透性,在结肠上皮细胞也能观察到超活化的Janus 激酶和 NF-κB,两者均与严重的结肠炎有关[15]。随着对 TRAIL 和免疫系统之间关系的不断研究,TRAIL 的另一个完全矛盾的角色渐渐被发掘,即 TRAIL 能损伤正常细胞。研究发现,慢性重症肝炎患者的外周 NK 细胞能够高表达 TRAIL,并对人正常的肝脏细胞产生毒性,同时患者的血清和肝脏中还存在丰富的炎性细胞因子(IL-6、IL-8),能够进一步增强 TRAIL 的表达和外周 NK 细胞的活化,因此在 IL-6/IL-8 的刺激和 NK 高表达 TRAIL 所介导的肝细胞毒性的情况下,正常的肝脏细胞对 TRAIL 诱导的凋亡越来越敏感,导致肝损伤[16-17]。
3 TRAIL 的耐药机制
3.1 TRAIL 功能性受体的缺失
TRAIL 耐药和 TRAIL 受体的缺失有密切关系,在多种类型的肿瘤细胞中均发现 DR4 和 DR5 功能性缺失。自噬体对 DRs 的吞噬作用导致 TRAIL 耐药。例如,在TRAIL 耐药的乳腺癌细胞 AU565 和 BT474 中,存在较高的自噬小体,DR4 和 DR5 共同连接在自噬体表面LC-3B上,导致膜表面 DR4/DR5 较少,从而产生耐药性。当使用 3-MA 破坏本底自噬体形成后,DR4/DR5 释放到膜表面,增强了 TRAIL 耐药细胞对 TRAIL 的敏感性。相反的,在 TRAIL 敏感的乳腺癌细胞 MDA-MB-231 中,膜表面 DR4/DR5 较高,并缺少本底自噬体,当使用巴弗洛霉素 A1 抑制溶酶体活性后,细胞中自噬体积累,使膜表面 DR4/DR5 含量降低,从而降低了细胞对 TRAIL 的敏感性[18]。通过转录调节和翻译后修饰调节能够调节 DRs 的表达与分布。转录因子 p53、CHOP、NF-κB、AP-1、FOXO3、Sp-1 等均能上调 DRs,而转录因子 GLI-3 和 YYI 分别下调 DR4、DR5。而某些癌细胞中,DRs 能在翻译后修饰过程中被自噬体、细胞核膜捕获,或者被保留在内质网/高尔基体中,导致细胞表面受体缺失,从而产生耐药。
此外,Ras GTPase 途径也能够调节 DRs 的表达。Ras家族包括 H-Ras、K-Ras、N-Ras,存在于 EGFR 的下游。在 TRAIL 耐药细胞中,只有 H-Ras 过表达,功能特异性地敲低 H-Ras 能够增加表面 DR4/5 的表达,增加 TRAIL耐药细胞对 TRAIL 的敏感性[11]。
3.2 对 TRAIL 诱导的凋亡途径的负性调节因子
3.2.1 拮抗受体——DcR1/DcR2/OPG 细胞内 DcR1、DcR2、OPG 受体表达较高时,能抑制 TRAIL 所诱导的凋亡途径,产生耐药。这三个拮抗受体因缺乏功能域 DD 展现出不同的抑制特性。OPG 是最弱的抑制剂,它与 TRAIL有较低的亲和力,在某种程度上表现出与 DR4/DR5 竞争性结合 TRAIL 来抑制凋亡。DcR1/DcR2 与 TRAIL 有较高的亲和力,DcR1 的 C 端有一个 GPI 结构,位于脂质筏,当 TRAIL 刺激时不能与 DR4/DR5 形成异源多聚体,因此,DcR1 也是通过竞争性结合 TRAIL 来抑制 TRAIL 诱导的凋亡;而 DcR2 能够在非脂质筏区域共同募集DR4/DR5,形成异源多聚体复合物,但 DcR2 缺乏 DD 功能域,因此它的募集通过空间阻碍来妨碍 caspase-8 的激活[3]。
3.2.2 sigma 1 受体 sigma 1 受体(Sig1R)是位于内质网膜的小分子跨膜蛋白,作为调节配体的分子伴侣,与TRAIL 诱导的凋亡有关。在前列腺癌细胞中,较高水平的Sig1R 能够使细胞对 TRAIL 产生更强的耐药性,因此,Sig1R 是 TRAIL 诱导凋亡的一个重要的调停蛋白[19]。
3.2.3 c-FLIP 同系物 在 TRAIL 耐药细胞中,细胞型Fas 相关死亡域样白介素 1β 转换酶抑制蛋白(celluar Fas-associated death domain-like IL-1β converting inhibitory protein,c-FLIP)过表达。c-FLIP 是 caspase-8/10 主要负性同系物,和 caspase-8/10 一样拥有两个 DED,能够被TRAIL DISC 募集,并且 c-FLIP 的长链形式包含一个caspase-like 的结构域,缺少 caspase 催化活性,能抑制caspase-8 链的形成、自我加工和激活,因此 c-FLIP 同系物主要抑制 TNF 家族诱导的凋亡。
3.2.4 Bcl-2 抗凋亡蛋白和凋亡抑制蛋白 在 II 型细胞中,Bax 的缺失或者 B 细胞淋巴瘤-2(B-cell lymphoma 2,Bcl-2)抗凋亡蛋白家族成员,如 Bcl-2、Mcl-1、Bc-xL 的过表达,均能抑制线粒体凋亡途径,产生耐药。而无论是I 型还是 II 型细胞,过表达的凋亡抑制蛋白(inhibitor ofapoptosis protein,IAP),如 XIAP、survive、cIAP,均能抑制 TRAIL 所诱导的凋亡。
3.3 生存信号途径对 TRAIL 诱导的凋亡的调节
TRAIL 启动的非凋亡途径包括 MAPK、NF-κB 和PI3K-PKB/Akt 等途径,均能增加不同种类正常/癌细胞增殖、生存、迁移、侵入或者炎症的能力,从而产生 TRAIL 耐药性。
TRAIL 激活的 MAPK 信号通路涉及一种可溶性的第二复合物的形成[3]。MAPK 主要包括三种不同的成员:胞外信号调节激酶(extracellular signal-regulated kinases,ERKs)、c-Jun 氨基末端激酶(c-Jun N-terminal protein kinases,JNKs)和 p38 裂原激活蛋白激酶(p38 mitogen-activated protein kinase,p38 MAP kinases)。ERKs 是第一个被发现与阻碍 TRAIL 诱导凋亡有关的 MAPK 成员,能抑制 ERK 途径,下调 Mcl-2 蛋白,使结肠癌细胞对 TRAIL 敏感。p38 MAP kinases 则被认为是调节酶的细胞因子。JNKs 涉及细胞增殖和凋亡,TRAIL 通过受体相互作用蛋白激酶 1(receptor-interacting protein kinase 1,RIPK1)和 TNF 受体相关因子 2(TNF receptor-associated factor 2,TRAF2)诱导 JNKs 途径。ERKs、JNKs 和 p38 的激活均依赖于细胞类型,并通过转录依赖或非依赖的方式调节 TRAIL 受体、c-FLIP、Bcl-2 家族蛋白的表达来影响TRAIL 诱导的凋亡。NF-κB 是核转录因子,在 TRAIL 耐药细胞已展现出 NF-κB 依赖性的炎症表型,增强 NF-κB信号能够抵抗 TRAIL 诱导的凋亡,提高肿瘤转移和侵入能力[4]。在 TRAIL 敏感细胞中,caspase-8 分裂受体相互作用蛋白 1(receptor interacting protein 1,RIP1)能封闭NF-κB 活性,进而启动 TRAIL 引起的凋亡,但如果将细胞持续暴露在低浓度的 TRAIL 中,激活的 NF-κB 不断被积累,从而增加 miR21/30C/100 产生,进一步下调caspase-8/TRAF-7/caspase-3/FoxO3a,导致 NF-κB 信号的增强,从而建立了一个正反馈环路,产生 TRAIL 耐药和上皮间质转化[20-21]。TRAIL 诱导激活 JNKs 和 NF-κB 途径分为两个阶段:早期阶段和延迟阶段。早期阶段是通过TRAF2/cIAP1 介导的 RIP1 泛素化进行激活,延迟阶段是通过 caspase 介导的 MEKK1 分裂和激活来产生的。cFLIP的过表达能够促进早期阶段但是抑制延迟阶段,当在 RIP1或者 TRAF2 缺陷的细胞中稳定地过表达 cFLIP,会发现细胞能够抵抗凋亡,但却无法激活 NF-κB。而 Bcl-2 的过表达对这两个阶段均产生促进作用。这些很好地解释了TRAIL 激活 JNK 和 NF-κB 途径的机制[22]。PI3K-PKB/Akt途径的激活也涉及 TRAIL 耐药。通过 PI3K 导致 Akt 的磷酸化和激活,而激活的 Akt 能磷酸化一系列底物,导致生存基因如 c-FLIP、Bcl-2、Mcl-1 或 XIAP 在转录水平上调或者磷酸化依赖性稳定,最终导致 TRAIL 耐药。在结肠癌细胞中抑制 PI3K/Akt 途径能使细胞对 TRAIL 敏感(图 3)。
4 TRAIL 联合用药策略
由于肿瘤细胞类型不同而存在多重 TRAIL 耐药机制。因此,TRAIL 联合用药的策略也具有多样性和针对性,而与 TRAIL 进行联合的药物或化合物的选择也是根据某种细胞的具体耐药机制进行针对性选择,大致可以分为以下几类:
⑴增加功能性受体 DR4/DR5 表达的药物或死亡受体激动剂抗体:如曲格列酮(troglitazone,TGZ)[23]。
⑵c-FLIP 抑制剂或者激活 DISC 水平的 caspase-8 的药物。
⑶Bcl-2、Mcl-1、Bc-xL 抑制剂或者 IAP 拮抗剂、survive 抑制剂。
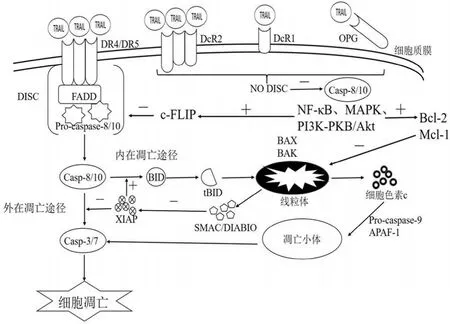
图3 TRAIL 的耐药机制
⑷组蛋白脱乙酰酶抑制剂(HDACi):如丙戊酸。
⑸与自噬相关的药物:如氯喹、姜辣素、橡黄素[8]。
⑹与抑制 NF-κB 和 MAPK 信号途径有关的药物:如化合物 YM155、海兔素(aplysin)。
⑺常规的化疗药物:如喜树碱、阿霉素、5-氟尿嘧啶、伊立替康、紫杉醇[23]。
⑻热休克蛋白 90 抑制剂:如 NVP-AUY922。
⑼其他:高热可以增强各癌症细胞系对 TRAIL 诱导的细胞死亡的敏感性,现已提出温和热休克治疗能通过引起c-FLIP 退化或激活线粒体凋亡途径来恢复 Fas 配体或TRAIL-induced 的凋亡[24]。
通常药物是通过多个途径同时抵抗耐药性,从而与TRAIL 产生联合抗肿瘤效果。例如,姜烯酚能够通过增加DR5 和促凋亡蛋白 Bax 表达,降低抗凋亡蛋白 survive 的表达来克服结肠癌细胞对 TRAIL 的耐药[25]。
5 前景
目前,对于 TRAIL 诱导细胞死亡及其耐药机制的研究已经取得了很大的进展,同时其联合用药的策略更是随着这些机制的完善而有了更多的发现,这些都将推动 TRAIL 通过临床研究。然而,由于肿瘤细胞的不同,TRAIL 耐药机制具有多样性,目前仍然无法寻找到一种或几种联合药物能广泛使用于肿瘤治疗中,并且 TRAIL 所介导的凋亡与自噬之间的复杂关系也需要更深入的研究去发现中间的关联机制。在未来,这些问题的解决将会加快 TRAIL 药物上市的步伐。
[1] Amarante-Mendes GP, Griffith TS. Therapeutic applications of TRAIL receptor agonists in cancer and beyond. Pharmacol Ther, 2015,155:117-131.
[2] Kao SY, Soares VY, Kristiansen AG, et al. Activation of TRAIL-DR5 pathway promotes sensorineural degeneration in the inner ear. Aging Cell, 2016, 15(2):301-308.
[3] Elmallah M, Micheau O. Marine drugs regulating apoptosis induced by tumor necrosis factor-related apoptosis-inducing ligand (TRAIL). Mar Drugs, 2015, 13(12):6884-6909.
[4] Hofmanová J, Straková N, Vaculová AH, et al. Interaction of dietary fatty acids with tumour necrosis factor family cytokines during colon inflammation and cancer. Mediators Inflamm, 2014, 2014:1-17.
[5] Lv S, Wang X, Zhang N, et al. Autophagy facilitates the development of resistance to the tumor necrosis factor superfamily member TRAIL in breast cancer. Int J Oncol, 2015, 46(3):1286-1294.
[6] Nazim UM, Jeong JK, Seol JW, et al. Inhibition of the autophagy flux by gingerol enhances TRAIL-induced tumor cell death. Oncol Rep,2015, 33(5):2331-2336.
[7] Kim SW, Lee JH, Moon JH, et al. Niacin alleviates TRAIL-mediated colon cancer cell death via autophagy flux activation. Oncotarget,2016, 7(4):4356-4368.
[8] Moon JH, Eo SK, Lee JH, et al. Quercetin-induced autophagy flux enhances TRAIL-mediated tumor cell death. Oncol Rep, 2015, 34(1):375-381.
[9] Xu J, Xu X, Shi S, et al. Autophagy-mediated degradation of IAPs and c-FLIP(L) potentiates apoptosis induced by combination of TRAIL and Chal-24. J Cell Biochem, 2016, 117(5):1136-1144.
[10] Jin SM, Jang HW, Sohn SY, et al. Role of autophagy in the resistance to tumour necrosis factor-related apoptosis-inducing ligand-induced apoptosis in papillary and anaplastic thyroid cancer cells. Endocrine,2014, 45(2):256-262.
[11] Twomey JD, Kim SR, Zhao L, et al. Spatial dynamics of TRAIL death receptors in cancer cells. Drug Resist Updat, 2015, 19:13-21.
[12] Singh K, Sharma A, Mir MC, et al. Autophagic flux determines cell death and survival in response to Apo2L/TRAIL (dulanermin). Mol Cancer, 2014, 13:70.
[13] Yang M, Liu L, Xie M, et al. Poly-ADP-ribosylation of HMGB1 regulates TNFSF10/TRAIL resistance through autophagy. Autophagy,2015, 11(2):214-224.
[14] Niu TK, Cheng Y, Ren X, et al. Interaction of Beclin 1 with survivin regulates sensitivity of human glioma cells to TRAIL-induced apoptosis. FEBS Lett, 2010, 584(16):3519-3524.
[15] Zhu J, Chen L, Shi J, et al. TRAIL receptor deficiency sensitizes mice to dextran sodium sulphate-induced colitis and colitis-associated carcinogenesis. Immunology, 2014, 141(2):211-221.
[16] Wan Z, Xie G, Wu Y, et al. Cytokines elevated in patients with HBV-related acute-on-chronic liver failure promote NK cell mediated cytotoxicity through TRAIL. Dig Liver Dis, 2016, 48(5):528-535.
[17] Netea-Maier RT, Plantinga TS, van de Veerdonk FL, et al. Modulation of inflammation by autophagy: Consequences for human disease. Autophagy, 2016, 12(2):245-260.
[18] Di X, Zhang G, Zhang Y, et al. Accumulation of autophagosomes in breast cancer cells induces TRAIL resistance through downregulation of surface expression of death receptors 4 and 5. Oncotarget, 2013,4(9):1349-1364.
[19] Das D, Persaud L, Dejoie J, et al. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) activates caspases in human prostate cancer cells through sigma 1 receptor. Biochem Biophys Res Commun, 2016, 470(2):319-323.
[20] Jeon YJ, Middleton J, Kim T, et al. A set of NF-κB-regulated microRNAs induces acquired TRAIL resistance in Lung cancer. Proc Natl Acad Sci U S A, 2015, 112(26):E3355-E3364.
[21] Zhang L, Dittmer MR, Blackwell K, et al. TRAIL activates JNK and NF-κB through RIP1-dependent and -independent pathways. Cell Signal, 2015, 27(2):306-314.
[22] Mitchell MJ, Wayne E, Rana K, et al. TRAIL-coated leukocytes that kill cancer cells in the circulation. Proc Natl Acad Sci U S A, 2014,111(3):930-935.
[23] Koyama M, Sowa Y, Horinaka M, et al. Peroxisome proliferator-activated receptor γ ligand troglitazone and TRAIL synergistically induce apoptosis. Oncol Rep, 2014, 31(2):947-954.
[24] Morlé A, Garrido C, Micheau O. Hyperthermia restores apoptosis induced by death receptors through aggregation-induced c-FLIP cytosolic depletion. Cell Death Dis, 2015, 6:e1633.
[25] Hwang JS, Lee HC, Oh SC, et al. Shogaol overcomes TRAIL resistance in colon cancer cells via inhibiting of survivin. Tumor Biol,2015, 36(11):8819-8829.
10.3969/j.issn.1673-713X.2016.05.011
国家自然科学基金(81373437、81321004)
100050 北京,中国医学科学院北京协和医学院医药生物技术研究所肿瘤室
陈淑珍,Email:bjcsz@imb.pumc.edu.cn
2016-07-12
猜你喜欢
青少年科技博览(中学版)(2022年11期)2023-01-07
保健医苑(2022年5期)2022-06-10
现代临床医学(2022年3期)2022-06-06
昆明医科大学学报(2022年2期)2022-03-29
昆明医科大学学报(2022年1期)2022-02-28
中老年保健(2021年3期)2021-12-03
汽车维修与保养(2021年8期)2021-02-16
科学大众(2020年12期)2020-08-13
世界农药(2019年2期)2019-07-13
医学研究杂志(2015年2期)2015-06-10
