
麻黄标准汤剂质量评价体系的建立
2017-04-06 16:39仝家羽赵嵘代云桃范自全王丹丹秦
中国中药杂志 2017年5期
仝家羽+赵嵘+代云桃+范自全+王丹丹+秦雪梅+陈士林
[摘要]建立麻黄标准汤剂质量控制方法,为所有中药饮片标准汤剂质量评价方法的制定提供参考。制备麻黄标准汤剂,建立UPLC-UV指纹图谱并测定其麻黄碱和伪麻黄碱总含量;采用UPLC-QTOF/MS对主要色谱峰进行结构确认,明确汤剂中的主要化学成分;计算出膏率、指标成分转移率、pH,评价工艺的稳定性。结果麻黄汤剂中麻黄碱与伪麻黄碱的平均总浓度为(2.11±0.70) g·L-1;所有麻黄水煎液指纹图谱相似度均大于0.85;麻黄标准汤剂对照指纹图谱主要共有峰有10个,包括生物碱、有机酸类和黄酮;麻黄标准汤剂出膏率为(17±3.2)%、麻黄碱与伪麻黄碱的整体转移率为(32.4±8.1)%。该文建立了系统评价麻黄标准汤剂的质量评价方法,为所有源于麻黄水煎液的制剂的质量控制标准的制定提供参考。
[关键词] 麻黄; 标准汤剂/标准煎液; 指纹图谱; 含量测定; 出膏率; 转移率
[Abstract] To establish the quality control methods for the standard decoction of Ephedrae Herba, and provide the reference for quality evaluation method of all Chinese herbal medicine decoction.Standard decoction of Ephedrae Herba was prepared, and UPLC-UV fingerprint was established to determine the total contents of ephedrine and pseudoephedrine. Then UPLC-QTOF/MS was used to confirm the major common peaks in the fingerprint to clarify the main chemical constituents in the decoction. In addition, the stability of the process was evaluated by calculating the parameters such as the extraction ratio, transfer rate of the index components and the pH values.In the decoction of Ephedrae Herba, the total average concentration of ephedrine and pseudoephedrine was (2.11±0.70) g·L-1; the similarities of all the fingerprints were more than 0.85; there were 10 major common peaks in the fingerprint, including alkaloids, flavonoids and organic acids; the extraction ratio was (17±3.2)%, and the overall transfer rate of ephedrine and pseudoephedrine was (32.4±8.1)%.The method for evaluating the quality of standard decoction of Ephedrae Herba was established in this article, providing reference for the quality control of products which were stemmed from the water extract of Ephedrae Herba.
[Key words] Ephedrae Herba; standard decoction; fingerprint; content determination; extraction ratio; transfer rate
湯剂作为中药传统的用药形式,至今仍被广泛使用,但由于其携带不便,煎煮耗时长和质量不均一等缺点给患者用药带来极大不便。为了解决以上问题,以汤剂为基础进行浓缩加工的中药配方颗粒等现代中药剂型不断出现。但由于缺乏统一的质量标准以及合理的中间监管过程造成了市场上现代剂型间剂量不统一、质量不稳定等问题[1]。2016年,陈士林研究员首次提出采用中药饮片标准汤剂来标化源于传统水煎液的不同用药形式,提高临床用药的准确性和疗效的一致性[2],中药标准汤剂,又称中药标准煎液,是经标准化工艺制备而成的单味中药饮片水煎液,用于标化临床用药,保障用药的准确性和剂量的一致性[2]。中药标准汤剂作为参照物,其质量标准的制定将为所有后续产品的质量标准的制定提供基础。
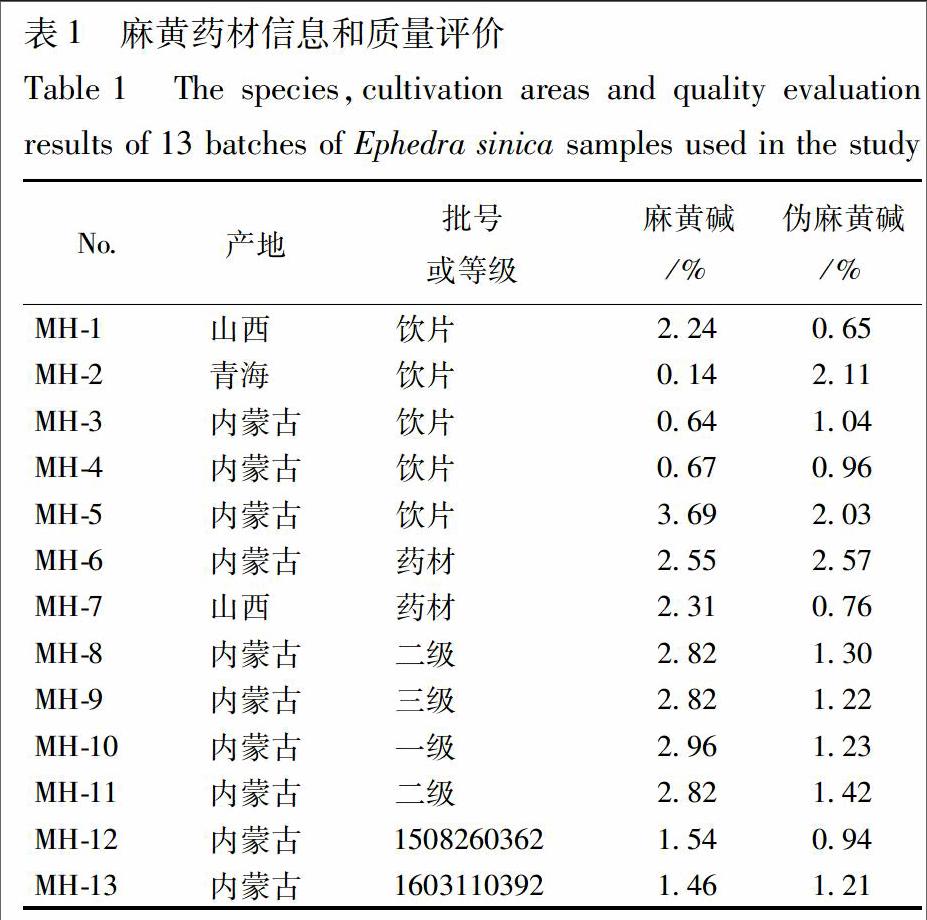
麻黄为麻黄科植物草麻黄Ephedra sinica Stapf 、中麻黄E. intermedia Schrenk et C. A. Mey .或木贼麻黄E. equisetina Bge.的干燥草质茎[3]。始载于《神农本草经》,列为中品。麻黄性温,味辛,微苦,有发汗解表,宣肺平喘,利水消肿的功效,其主要化学成分为生物碱、挥发油,同时含有少量的黄酮、多糖和有机酸类等,目前一般认为其主要有效成分是生物碱和挥发油类[4]。相较于2015年版药典中规定的以麻黄碱和伪麻黄碱的总量判断药材是否合格,中药指纹图谱更能整体性地反映药材质量,但目前对于麻黄指纹图谱的研究均为酸提或有机试剂提取,这与中药水煎的传统用药方式还存在着较大的差别。
本文按照《中药饮片标准汤剂研究策略》中推荐的制法[2]制备了麻黄标准汤剂,建立了其质量评价方法。《中国药典》规定的3种麻黄间差距较大,现选取市场上商品药材量最大的草麻黄为研究对象制备汤液[5],对主要工艺参数出膏率范围、指标成分转移率范围和pH进行了标定;采用药典方法对麻黄汤剂中主要药效成分麻黄碱和伪麻黄碱进行了含量测定;建立了标准汤剂对照指纹图谱,采用对照品比对和液质联用鉴定,对指纹图谱的主要共有峰进行指认,共指认了8个成分。本文展示了对麻黄标准汤剂从源头药材质量控制、中间过程参数标定和标准汤剂的化学指纹标定的整个质量控制过程,为麻黄饮片标准汤剂的研究提供参考。
1 材料
Acquity UPLC H-class超高效液相系统,Waters Xevo G2-XS QTOF质谱系统(Waters Corporation,Milford,MA,USA),Unifi 1.8软件。水为娃哈哈纯净水,乙腈、甲醇为色谱纯,其余试剂均为分析纯。三爱思pH精密试纸。麻黄碱、伪麻黄碱均购于北京世纪奥科生物技术有限公司,纯度为99.9%。麻黄药材共13批,购于内蒙古、山西、青海、安徽亳州市场等地,包括了麻黄的主产区,道地产区和国内的主要的药材市场;DNA测序与数据库比对鉴定为麻黄科植物草麻黄E. sinica的干燥草质茎。根据《中国药典》2015年版麻黄的含量测定项测定麻黄碱和伪麻黄碱的含量,见表1,结果显示所有批次中麻黄碱与伪麻黄碱的总含量均大于0.8%,药材全部符合药典标准。
2 方法与结果
2.1 供试品溶液的制备
2.1.1 麻黄标准汤剂制备方法
称取麻黄饮片100 g,置于圆底烧瓶中,加12倍水,充分润湿,放置浸泡30 min,加热煮沸后回流提取30 min,趁热3层纱布过滤,滤渣再加入10倍水回流提取20 min,3层纱布滤过,合并滤液并水浴浓缩至500 mL即得。
2.1.2 指纹图谱供试品溶液的制备
取2.1.1项下所得的麻黃标准汤剂置于2 mL离心管中,12 000 r·min-1离心5 min,取上清液即得。
2.1.3 含量测定供试品溶液的制备
取2.1.1项下所得的麻黄标准汤剂0.4 mL,置于离心管中,加水稀释至1 mL,12 000 r·min-1离心5 min,取上清液即得。
2.1.4 对照品溶液的制备
取麻黄碱和伪麻黄碱对照品适量,精密称定,置棕色量瓶中,加甲醇制成麻黄碱与伪麻黄碱质量浓度均为60 g·L-1的溶液,摇匀,作为对照品储备液。
2.2 HPLC含量测定
2.2.1 HPLC色谱条件
采用Welch Materials HPLC Phenyl-Ether柱(4.6 mm×250 mm,5 μm);柱温为30 ℃,流速1 mL·min-1;进样量10 μL;流动相0.092%磷酸+0.04%三乙胺+0.02%二正丁胺(A)-甲醇(B),A-B 98.5∶1.5等度洗脱;检测波长210 nm。
2.2.2 HPLC分析方法学考察
2.2.2.1 线性关系考察 将2.1.4项下混合对照品储备液,分别用甲醇稀释为质量浓度分别是2.5,10,20,40,50,60 g·L-1的溶液,按2.2.1项下的色谱条件测定,进样10 μL,以进样量10 μL中对照品质量(mg)为横坐标,210 nm波长下的峰面积为纵坐标,绘制标准曲线。麻黄碱的线性方程为y=15 435x+25.421,r=0.999 9,伪麻黄碱的线性方程为y =16 995x+56.849,r=0.999 8。
2.2.2.2 精密度 取供试品溶液按HPLC色谱条件进样6次,每次进样量为10 μL,麻黄碱峰面积RSD为0.12%,仪器精密度良好,符合含量测定要求。
2.2.2.3 稳定性 取供试品溶液放置0,1,4,6,12,24 h后按HPLC色谱条件进样10 μL进行测定,记录所有共有峰的峰面积,24 h内麻黄碱峰面积RSD为0.67%,说明麻黄供试液在24 h内稳定,符合含量测定要求。
2.2.2.4 重复性 取同一批样品,按供试品备样方法平行制备6份供试品溶液,分别按HPLC色谱条件进样10 μL进行测定。麻黄碱峰面积RSD为0.60%,说明本实验采用的方法重复性良好。
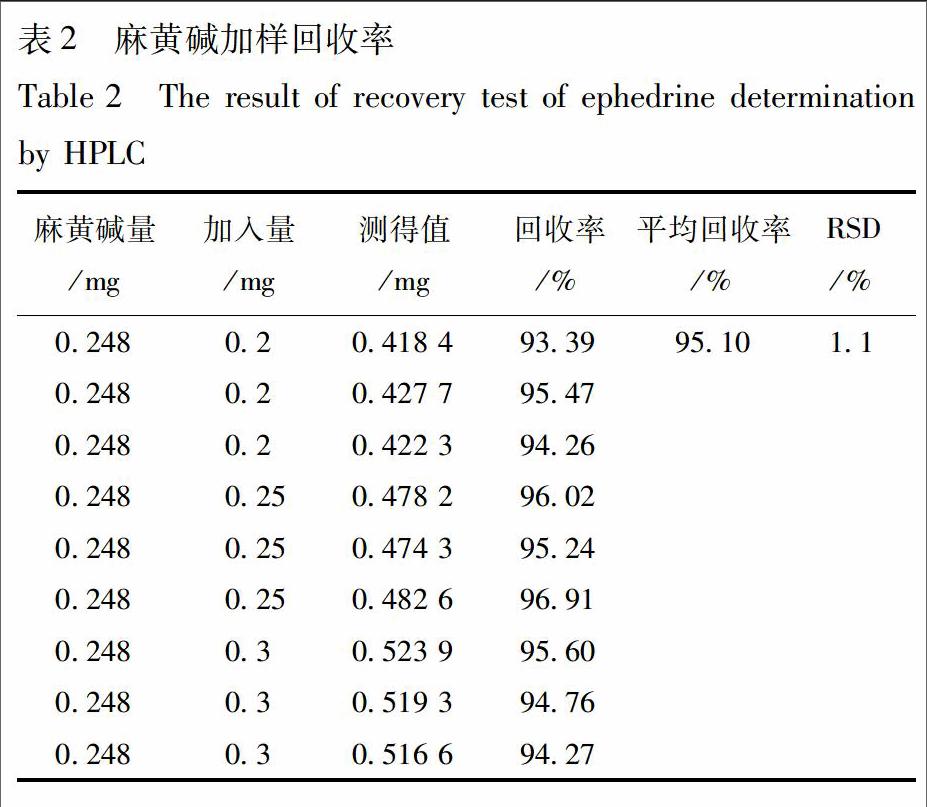
2.2.2.5 回收率 平行精密量取1 mL已知麻黄碱质量分数为0.248 g·L-1的标准汤剂溶液9份,分别加入麻黄碱对照品0.2,0.25,0.3 mg各3份,混匀,按照供试品备样方法备样,进样10 μL,记录麻黄碱峰面积,计算含量及回收率,结果见表2,结果显示其平均回收率为95.10%,RSD 1.1%,表明该方法可以用于麻黄碱的准确测定。
2.2.3 HPLC含量测定
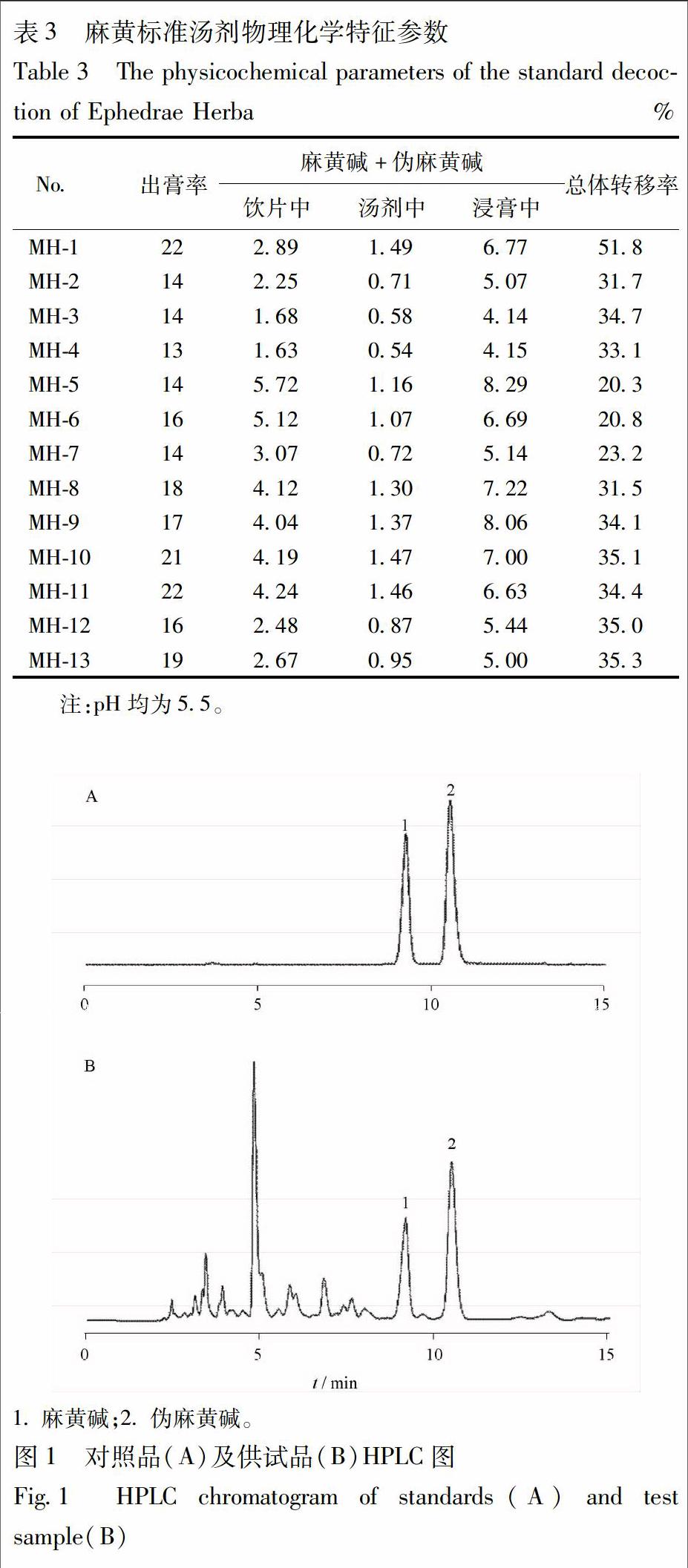
分别精密吸取13批供试品溶液10 μL,注入液相色谱仪,按2.2.1项下的色谱条件测定,记录210 nm波长下麻黄碱与伪麻黄碱的色谱峰面积,带入标准曲线进行计算,结果见表3。对照品及供试品HPLC液相色谱图见图1,麻黄中的主要成分是麻黄碱与伪麻黄碱,两者结合共同发挥药效,其总量更能体现药材质量。13批麻黄(MH1~MH13)的标准汤剂中,麻黄碱与伪麻黄碱总量的质量分数为1.08~2.98 g·L-1,均值为(2.11±0.70)g·L-1;根据各批药材不同的出膏率,浸膏粉中麻黄碱与伪麻黄碱总量的质量分数为4.14%~8.29%,平均值为(6.12±1.39)%。
2.3 UPLC指纹图谱测定及共有峰鉴定
2.3.1 UPLC色谱条件及质谱条件
采用Acquity UPLC CSH C18柱(2.1 mm×100 mm,1.7 μm,Waters);柱温为40 ℃;流速0.4 mL·min-1;进样量为1 μL;流动相0.1%甲酸+0.1%三乙胺水(A)-乙腈(B),梯度洗脱(0~0.5 min,0.5%~0.5% B;0.5~15 min,0.5%~15% B;15~20 min,15%~30% B;20~25 min,30%~99.5% B);检测波长260,210 nm。采用Xevo G2-XS QTOF质谱仪,电喷雾电离离子源 (ESI),离子化模式为正、负离子,离子源温度为150 ℃,脱溶剂气体为高纯度氮气,温度为550 ℃,流速为800 L·h-1,毛细管电压为1.0 kV,锥孔电压为30 V,扫描范围m/z 50~1 200。亮氨酸-脑啡肽(m/z 554.261 5)作为外标(Lock SprayTM)进行质量实时校正。
2.3.2 UPLC指紋图谱采集与分析
分别精密吸取13批供试品溶液1 μL,注入超高效液相色谱仪,按2.3.1项下的色谱条件测定,得13批麻黄提取物210,260 nm波长下的UPLC指纹图谱。210 nm波长下谱图杂乱,共有峰少,但主要药效成分麻黄碱与伪麻黄碱信号明显,260 nm波长下谱图较为平整,共有峰多且均匀分散,考虑到麻黄碱与伪麻黄碱已通过含量测定的手段加以监控,故选择更能体现整体药材质量的260 nm波长下的图谱作为麻黄的指纹图谱。图谱采用药典委推荐的“中药色谱指纹图谱相似度评价系统(2012)”软件进行色谱峰匹配,计算谱图的相似度;找出13批药材的共有峰,计算其相对保留时间、峰面积、峰面积百分含量、相对峰面积。13批麻黄提取物260 nm波长下的指纹图谱匹配结果见图2,相似度在0.86~0.98,共找出10
个峰面积百分比大于2%、峰形较好、稳定且易辨识的共有峰。以5号甲基麻黄碱峰作参照,计算10个共有峰的保留时间、相对保留时间、峰面积、峰面积百分比、相对峰面积,见表4。
2.3.3 共有峰指认
吸取2.1.1供试品溶液1 μL,注入UPLC-QTOF-MS系统,按2.3.1项下的色谱条件运行,记录质谱信号。采用Mass Lynx 4.1 对正负离子模式总离子流图进行处理,采用UNIFI 1.8 数据处理系统,通过比对精确相对分子质量、特征碎片峰,并结合Mass FragmentTM确定化合物的相对分子质量和可能分子式,比对已建立的目标数据库和文献[6-8],推测260 nm波长下共有峰可能的化合物结构,UV及质谱总离子流图见图3,相关离子推断见表5,共鉴定出8个共有峰,2个给出分子式。
2.4 麻黄标准汤剂过程稳定性评价指标参数的测定
2.4.1 出膏率
分别取2.1.1项下的供试品溶液50 mL,干燥,称取浸膏质量(m),根据如下公式计算标准汤剂的出膏率,出膏率=干膏量/饮片量×100%=m×10/100×100%。结果见表3,13批煎液出膏率为13%~22%,平均值为(17±3.2)%,不同批次的出膏率相差不大。
2.4.2 转移率
分别将麻黄汤剂和麻黄饮片中麻黄碱和伪麻黄碱的含量总和带入如下公式计算标准汤剂的转移率,转移率=汤剂中指标成分量/饮片中
指标成分量×100%。结果见表3,转移率以麻黄碱与伪麻黄碱总量计算为20.3%~51.8%,平均转移率为(32.4±8.1)%,相对稳定。
2.4.3 pH
将pH精密试纸浸入麻黄溶液中,0.5 s后取出与标准色版比较,即得pH。平行测定3次,取平均值。结果见表3,pH 5.5,不同批次之间没有差异。
3 讨论
3.1 质量控制方法
本文对麻黄标准汤剂的质量控制主要从三方面把关:①药材选择范围广,包括了麻黄的主产区和主要的药材市场,共13个批次,对药材鉴定精确到物种,实验前,对药材进行了检测,符合2015年版《中国药典》各项规定的进入下一步实验;②标准汤剂的整个制备过程都完全遵守统一的标准化操作[1];③标准汤剂的质量控制,采用化学指纹图谱和指标成分含量测定相结合的模式,鉴定了8个主要共有峰对应的化学成分,从整体定性和指标成分定量的角度标定麻黄汤剂的化学轮廓和含量标准。
3.2 对比日本汉方药优势
已有的日本汉方药也是从含测、出膏率、转移率、煎煮工艺来达到质量控制,但日本用药习惯与国内有很大差距[9-10],主要是:①用药量较国内小,一般在20 g左右;②药材煎煮前需要粉碎,而非饮片直接投料;③加水量多,为药材质量的20倍,只煎1次。相较与日本汉方药的质量控制,本文的质量规范更符合中医理论及临床用药习惯,各个过程的标准更加细化,更有利于质量及临床疗效的均一化。除此之外,本文加入了指纹图谱和共有峰鉴定,从整体定性角度为麻黄质量标准的制定提供参考,更符合中药倡导的整体性理念。
3.3 指标成分选择
麻黄标准汤剂中指标成分的定量上,参考2015年版《中国药典》,将麻黄碱与伪麻黄碱总量作为指标,结果显示该方法可用于水煎液中麻黄碱与伪麻黄碱的测定。水煎液中麻黄碱和伪麻黄碱浓度总和为1.08~2.98 g·L-1,变化范围位于均值的52%~141%;而水煎液中麻黄碱质量浓度为 0.14~3.69 g·L-1,变化范围位于均值的7%~180%;伪麻黄碱质量浓度为0.65~2.57 g·L-1,变化范围位于均值的49%~192%。该数据表明,二者之和的变化范围远小于单个成分的变化范围,该现象
可能与加热煎煮过程中二者的相互转化有关。现代药理学一般认为麻黄碱与伪麻黄碱药效相似,平喘中以麻黄碱为主,伪麻黄碱为辅[11-12]。因此,从二者可能的结构转化和药效角度考虑,指标成分选择中采用二者之和更为合适。
3.4 色谱峰指认
本文首次报道了麻黄标准汤剂的指纹图谱,并对主要的色谱峰进行了成分指认。麻黄标准汤剂260 nm下的共有峰的质谱鉴定结果显示,主要色谱峰包括1个生物碱成分(甲基麻黄碱),1个有机酸[5-(β-D-吡喃葡萄糖氧基)-2-羟基苯甲酸]及多个黄酮类成分(文赛宁-2、夏佛托苷、芹菜苷、佛来心苷)。已有文献报道麻黄中的主要成分[4]包括生物碱类、黄酮类、有机酸类、挥发油类等。结合质谱色谱图,可以看出260 nm检测出麻黄标准汤剂的主要小分子化学成分,呈现了除麻黄碱和伪麻黄碱外的麻黄汤剂中的主要成分。
3.5 檢测波长的选择
指纹图谱是控制标准汤剂、配方颗粒等失去中药外表特征的中药制剂的有效手段。标准汤剂指纹图谱中,液相检测波长的选择要能稳定反应药材中的主要成分。麻黄中生物碱成分与其他成分性质差距较大,受酸碱度影响波动大,难以在一张图谱中同时呈现。本实验采集了对生物碱类成分有最大吸收的210,260 nm的图谱,结果显示210 nm下主要药效成分麻黄碱与伪麻黄碱信号明显,现有文献正是因为这个原因,指纹图谱波长均在210 nm附近,但其谱图杂乱,共有峰少;260 nm下的图谱优于210 nm,谱图更为平整,共有峰多且均匀分散;因此,质量标准中采用2个主要生物碱成分的定量标化和260 nm波长下的指纹图谱相结合的模式,该方法能全面反映、定量和定性麻黄中的主要成分。
4 总结
采用本文建立的方法,所得麻黄标准汤剂的平均出膏率为(17±3.2)%,出膏率为13%~22%,位于均值的75%~130%;麻黄标准汤剂中麻黄碱与伪麻黄碱的总体平均转移率为(32.4±8.1)%,转移率为20.3%~51.8%,位于均值的63%~160%;浸膏粉中麻黄碱与伪麻黄碱的总体质量分数为(6.12±1.39)%,范围是4.14%~8.06%,位于均值的68%~132%。pH均为5.5,各样本间无差异。这些数据的变化范围均在60%~160%。13批麻黄标准汤剂相似度值位于0.86~0.98,8个主要共有峰的峰面积百分含量和相对峰面积批间差距也较大,其原因可能与麻黄化学成分复杂,共有峰面积百分比较小,药材个体间差异较大而导致细小的非共有峰较多有关。鉴于麻黄个体间的较大差异因此,建议麻黄标准汤剂质量标准的制定要适度放宽范围。
[参考文献]
[1] 张红梅,宋景政,谭红胜,等.从汤剂到颗粒剂:中药配方颗粒20年回顾与展望[J].世界科学技术——中医药现代化,2012(4):1740.
[2] 陈士林,刘安,李琦,等.中药饮片标准汤剂研究策略[J].中国中药杂志,2016,41(8):1367.
[3] 中国药典.一部[S]. 2015:320.
[4] 赵巍.草麻黄化学成分研究[D].北京:中国协和医科大学,2009.
[5] 洪浩,陈虎彪,徐风,等.麻黄药材原植物资源和市场品种调查[J].中国中药杂志,2011,36(9):1129.
[6] Lv M,Sun J,Min W,et al.GC-MS based metabolomics study of stems and roots of Ephedra sinica[J].J Pharm Biomed Anal,2015,114:49.
[7] 罗佳波,李吉来,陈飞龙,等.麻黄汤中化学成分的GC-MS分析[J].中国实验方剂学杂志,2001(1):1.
[8] 孟翔宇,宋凤瑞,刘志强,等.麻黄中主要化合物的串联质谱研究[J].质谱学报,2006,27(suppl):55.
[9] 董丽丽,李野,刘春波.日本汉方药发展概况及其借鉴意义[J].国际医药卫生导报,2004(13):66.
[10] 郭晓,郁洋.日本汉方药的发展及对我国中药产业的启示[J].亚太传统医药,2007(9):9.
[11] 刘赜,石倩,杨洋,等.麻黄碱与伪麻黄碱平喘效果及机制比较研究[J].中草药,2009(5):771.
[12] 卫平,郑芳昊,霍慧灵,等.不同配伍比例对麻黄-甘草药对中麻黄类生物碱成分血浆药动学的影响[J].中国实验方剂学杂志,2016,22(7):100.
[责任编辑 孔晶晶]
猜你喜欢
现代临床医学(2021年1期)2021-01-26
通化师范学院学报(2020年12期)2020-12-21
中成药(2017年6期)2017-06-13
考试周刊(2016年95期)2016-12-21
中国烟草学报(2012年1期)2012-04-09
