
B972-PFD:一种高精度的色散校正密度泛函方法
2017-06-21 12:33王一波
物理化学学报 2017年6期
何 禹 王一波,*
(1贵州省高性能计算化学重点实验室,贵阳 550025;2贵州大学网络与信息中心,贵阳 550025)
B972-PFD:一种高精度的色散校正密度泛函方法
何 禹1,2王一波1,2,*
(1贵州省高性能计算化学重点实验室,贵阳 550025;2贵州大学网络与信息中心,贵阳 550025)
发现一种与球原子经验色散模型SAM深度契合的杂化泛函B972,组合成高精度的色散校正密度泛函B972-PFD。采用S66、S66x8和S22标准数据集以及大气氢键团簇、Adenine-Thymine的π…π堆叠、Watson-Crick氢键复合物和甲烷结合(H2O)20水簇等体系测试了B972-PFD的性能。测试结果显示:对于S66数据集B972-PFD方法的精度与Head-Gordon研究组的三个新泛函ωB97X-V、B97M-V和ωB97M-V处于同一水平,相对于CCSD(T)/CBS金质标准,结合能的RMSD小于1 kJ·mol-1;在其它数据集的测试中,B972-PFD方法也表现出很好的计算精度。通过研究基函数效应,我们推荐Pople的6-311++G(2d,p)作为B972-PFD方法的最优性价比基组。
分子间相互作用;密度泛函色散校正;球原子色散模型;B972-PFD
1 引 言
在分子间相互作用能计算中,由于常见的近似密度泛函方法缺乏对长程色散作用的有效描述,迄今为止,对于Hobza的分子间相互作用标准测试集S661,以CCSD(T)/CBS结果为标准,不借助于各类色散校正,还没有一种仅使用电子密度分布及其导数的泛函能够独立地把分子间相互作用能均方根误差(RMSD)计算到低于1 kJ·mol-1水平。2005年至2016年间,Truhlar研究组一直致力于发展这一类密度泛函—Minnesota泛函,近期报道了其最新的泛函MN152,随后Head-Gordon研究组很快发表了对14种Minnesota泛函的详细评测工作3,结果表明使用足够大的基函数def2-QZVPPD,对于S66数据集该系列泛函的RMSD误差在1.38-9.04 kJ·mol-1之间,表现最好的是M06-2X泛函。
密度泛函结合二级微扰理论MP2的双杂化泛函方法取得了重要进展,对弱相互作用的描述已逐渐精确。我们的测试表明,早期双杂化泛函B2PLYP4的S66数据集RMSD达到5.76 kJ·mol-1,后来Chai研究组提出的PBE0-25和ωB97X-26双杂化泛函,RMSD减小到3.40和1.77 kJ·mol-1,Xu与Goddard研究组发展的另一类双杂化泛函XYG37,XYGJ-OS8,RMSD减小为1.61和1.79 kJ·mol-1。
近年来,密度泛函的各类色散校正方法发展迅速9,Grimme的经验色散校正DFT-D3方法及参数集支持94种元素和常见的几十种泛函10,S66测试集的RMSD误差在1.35-1.88 kJ·mol-1之间,表现最好的是B3PW91-D3(BJ)为1.35 kJ·mol-1;Head-Gordon研究组基于VV10非局域(Non-Local)校正11发展了另一类ωB97X-V12、B97M-V13和ωB97M-V14泛函,是目前计算分子间相互作用能最为成功的方法,它们把S66测试集的RMSD误差减小到1 kJ·mol-1以内14,见图1与表S1 (见Supporting Information (SI))。
还有一类基于球原子模型(spherical atom model)的色散校正方法,也称Petersson-Frisch色
散校正(Petersson-Frisch dispersion),已被Petersson、Frisch等结合APF泛函作为APF-PFD标准方法15加入到Gaussian 09和Gaussian 16程序中16,17。SAM色散校正模型物理图像清晰、简单明了,它不同于Grimme的DFT-D3模型,除了原子预先用标准方法计算的HOMO能量和极化率物性数据外,没有针对元素,没有依赖于泛函参数化,也没有加入到Kohn-Sham方程能量算符中参与自洽场迭代、不依赖于基函数,是一类值得关注、并具有较好应用前景的色散校正方法(SAM色散校正算法的简单描述见SI Part 2)。正是由于SAM色散校正模型的这些特性,使得它对泛函的选择性很强,其应用关键是找到与之契合的泛函。这类研究中,APF泛函并非很成功,此前我们发现它与SAM结合构成的APF-PFD方法过高地估计了色散作用,通过适度地减小SAM色散能贡献,提出了改进的APF-D*方法或称APF-PFD*,提高了一般性分子间相互作用问题结合能计算精度18。另外一种思路是不改变SAM色散能,能否寻找到一种泛函与其更好地契合,获得高精度的结合能?为此我们对15种常见密度泛函进行了SAM色散模型的匹配筛选,最终发现B97219是与SAM模型契合得最好的泛函,B972-PFD/6-311++G(2d,p)的S66测试集RMSD从APF-PFD及APF-PFD*的3.27、1.76下降到0.96 kJ·mol-1,达到低于1 kJ·mol-1的高精度水平。
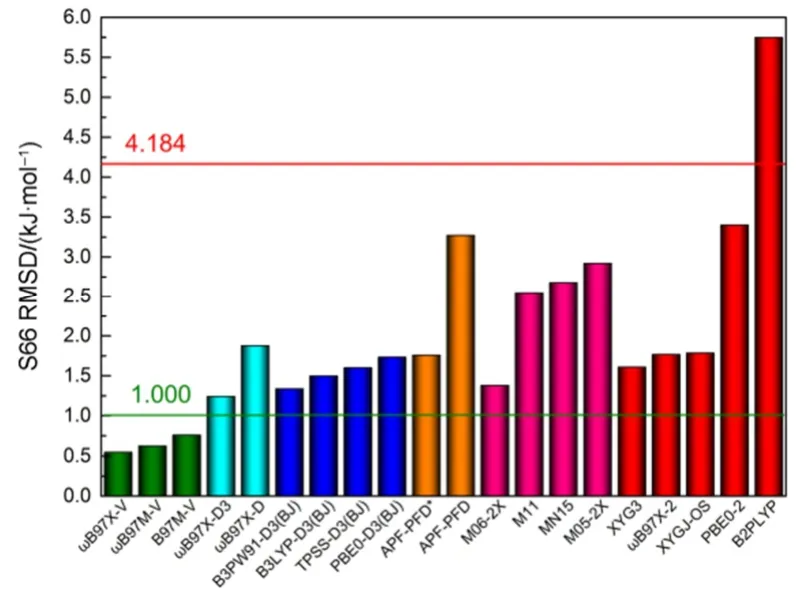
图1 S66测试集各类泛函计算精度范围Fig.1 Comparison of calculation precision for different DFT methods on the S66 test set
2 方法与计算
2.1 DFT方法筛选
DFT-D方法计算结果的准确性除了DFT泛函和色散校正模型自身的品质外,很大程度取决于两者对色散作用描述的互补性或契合程度。为构造与SAM球原子色散模型相适应的泛函,Petersson、Frisch等将41.1% B3PW91和58.9% PBE0 混合成APF泛函,但是APF泛函并没有达到作者预期的效果18。计算单分子热力学性质较成功的泛函通常对短程和中程色散的描述是充分的,不一定需要重新构造和优化泛函。因此,从常用的B3LYP20、BLYP21、PBE22、PBE023、B3PW9124、TPSS25、ωB9726、ωB97X26、APF15、B9727、B97128、B97219、B97329、B97-K30和X3LYP31等15种泛函中寻找与SAM色散模型相契合的泛函,期望获得高精度的结合能计算结果。
2.2 基函数效应
发现目标泛函后,在校正基函数重叠误差BSSE的前提下,研究这种泛函的基函数效应,从6-311++G(idjf, kpld)(i = 2, 3, j = 0, 1, k = 1, 2, 3, l = 0, 1)、cc-pVnZ、aug-cc-pVnZ (n = D, T, Q)32-34、def2-TZVP35、def2-TZVPP35、def2-QZVPP35等15种常见基组中,研究基组收敛性,寻找出具有较高性价比的基函数。
2.3 训练与测试数据集
Hobza研究组提出的S66是被广泛使用的分子间相互作用数据集,我们选择其作为本文方法探索的训练集。它由66对源于有机、生物体系的二聚体构成,各类分子间相互作用类型都均衡地包含在其中,对静电和色散作用的考虑较为适度,氢键组选择了23对典型的不同强度和类型的以静电作用为主的氢键复合物,色散组选择了23对涵盖π…π,C―H…π和C―H…C―H等以色散作用为主的复合物,混合组选择了20对静电与色散并存的复合物。其中各复合物的几何结构数据使用校正BSSE的MP2/cc-pVTZ方法优化得到,结合能数据以CCSD(T)/CBS金质标准的结果为基准。
当确定与SAM模型最优契合的目标泛函后,我们选取下列数据集进一步测试其性能:
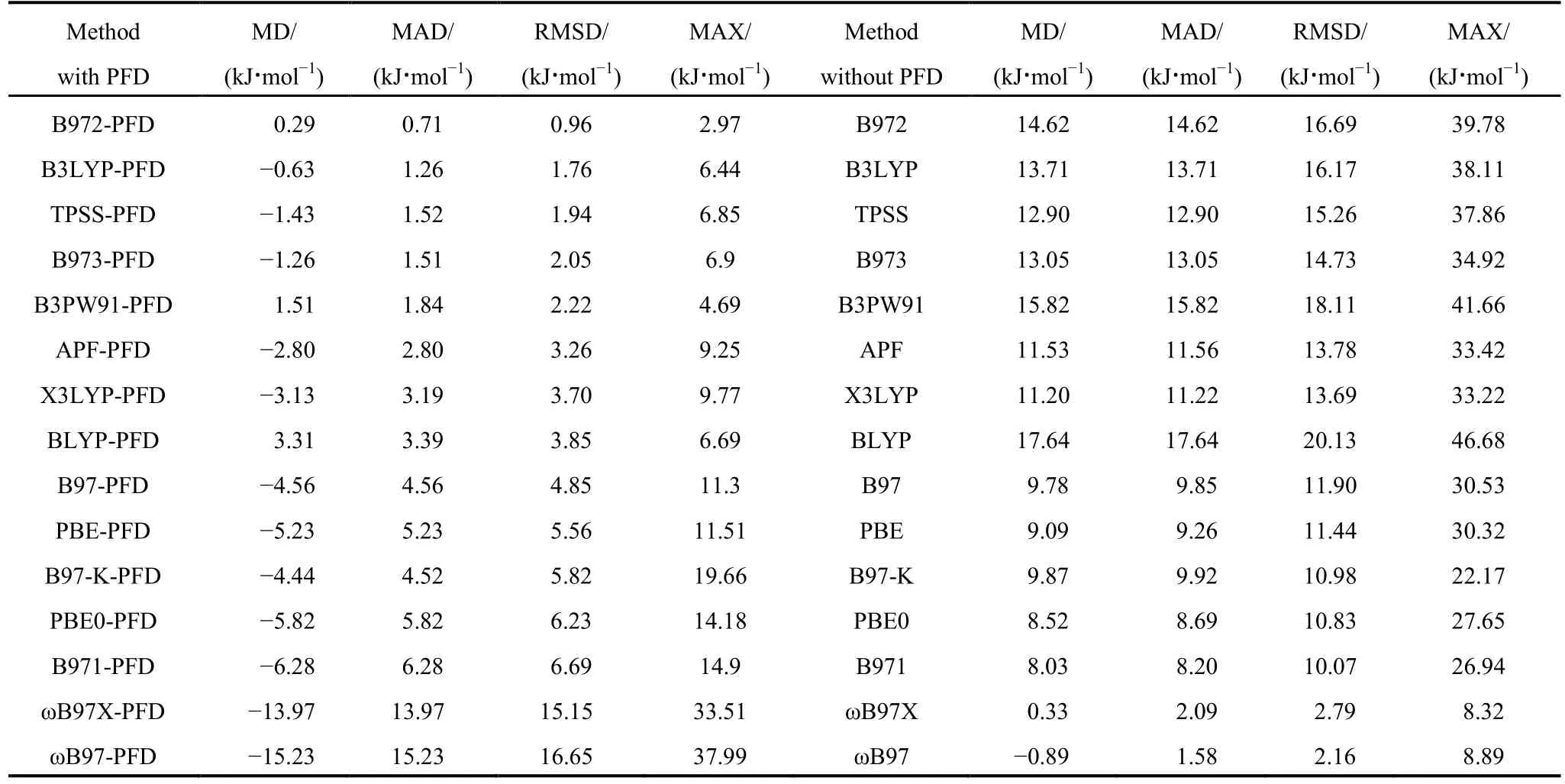
表1 筛选泛函DFT-PFD/6-311++G(2d,p)和DFT/6-311++G(2d,p)以CCSD(T)/CBS为基准的S66数据集误差分析Table 1 Calculated errors of DFT-PFD/6-311++G(2d,p) and DFT/6-311++G(2d,p) methods with respect to the benchmark CCSD(T)/CBS calculations on the S66 data set
第一,S22数据集。它也是Hobza研究组早于S66提出的分子间相互作用数据集36,由22对常见非键相互作用类型的二聚体构成,尽管其中一部分与S66有重复,但在早期应用更加广泛,是平衡结构下,训练、测试和评价非键相互作用理论方法的经典数据集。
第二,大气化学强氢键数据集。硫酸与胺、有机酸氢键簇合物的研究是探索大气气溶胶颗粒形成机制的基础,迄今为止已有不少实验与理论工作发表,最近Elm和Kristensen选取了H2SO4与H2O、NH3、CH3NH2、(CH3)2NH、(CH3)3N、H2N(CH3)2NH2、HCOOH、CH3COOH、HCO3H、CH3CO3H、H2SO4等11种二元复合物,使用收敛的基函数,建立了迄今为止这类体系最为精确的CCSD(T)/CBS能量标准值37。近年来密度泛函借助于各类校正方法,大大提升了过去单纯泛函计算不是很成功的色散型复合物能量计算精度,而这些校正方法,特别是色散校正,对氢键复合物计算精度的提高,效果远不及色散型复合物。要获得各类氢键键能的精确计算结果对密度泛函方法仍然是挑战。用此数据集的目的是测试目标泛函在伴随质子迁移的强氢键体系的性能,探索其在大气环境化学领域计算的应用价值。
第三,S66x8数据集。它也源于Hobza研究组38,是非平衡构型下的数据集,其固定S66中各单体结构,将两单体间距离分别调整到平衡位置的0.90、0.95、1.00、1.05、1.10、1.25、1.50、2.00倍,共528个测试点,旨在测试目标泛函势能面扫描、几何结构优化的性能。
2.4 数据统计分析指标
对于各种方法与参考标准差别的统计分析,使用了平均偏差MD、平均绝对偏差MAD、最大偏差MAX与均方根偏差RMSD统计指标(定义见SI Part 3)。其中RMSD最为重要,反映了数据偏差的离散程度,MD与MAD反映了系统的误差,通常综合RMSD与MAD指标进行整体评价,MD与MAX指标辅助参考。
2.5 计算过程
本项研究主要计算工作采用Gaussian 09 Rev. D01程序16,其它涉及ωB97X-V、B97M-V、ωB97M-V和ωB97X-D3泛函、XYG3、XYGJ-OS、PBE0-2和ωB97X-2双杂化泛函等的计算使用Q-Chem 4.4.1程序39,在贵州大学云计算平台Dell PowerEdge R210 Linux集群上完成。
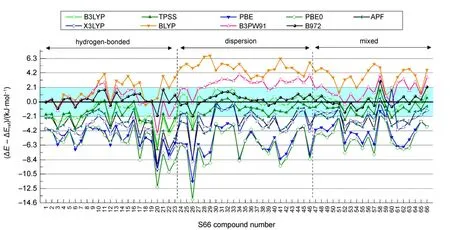
图2 筛选泛函DFT-PFD/6-311++G(2d,p)以CCSD(T)/CBS为基准的S66数据集误差分析Fig.2 Calculated errors of DFT-PFD/6-311++G(2d,p) methods with respect to the benchmark CCSD(T)/CBS calculations on the S66 data set
3 结果及讨论
3.1 筛选与SAM色散模型契合的密度泛函
选择单分子化学性质研究常用的B3LYP、BLYP、PBE、PBE0、B3PW91、TPSS、ωB97、ωB97X、APF、B97、B971、B972、B973、B97-K和X3LYP等15种泛函,使用6-311++G(2d,p)基分别计算了S66数据集各复合物使用或不使用SAM色散矫正的结合能,以CCSD(T)/CBS为基准计算出各复合物结合能的误差,并统计MD、MAD、RMSD与MAX值,结果见表1和SI表S2,S3及图2,3。
分析上述结果不难发现,对于S66数据集,与CCSD(T)/CBS基准值比较,在被筛选的15种泛函中,B972是与SAM色散模型契合得最好的泛函,加上SAM色散校正后,其RMSD最小为0.96 kJ·mol-1,MD、MAD和MAX也最小。从图2可看出,BLYP与B3PW91泛函,自身色散描述
不足,仅靠SAM色散校正,结果偏小;APF、X3LYP、B3LYP、TPSS、PBE与PBE0的误差出现在零基准线的下方,这些泛函自身计算出的色散能加上SAM色散校正后,都过高估计了色散作用,B3LYP的RMSD为1.76 kJ·mol-1,TPSS为1.94 kJ·mol-1,PBE和PBE0高估的幅度较大,RMSD分别为5.56和6.23 kJ·mol-1,特别是第20、26号复合物误差分别超过-12及-14 kJ·mol-1,这并不难理解,过去在没有任何色散校正的情况下,PBE和PBE0泛函也常用于分子间相互作用的研究40-43;另外,SAM色散校正较好地补偿了X3LYP泛函对色散作用为主的体系色散能描述的不足,但是对于氢键体系X3LYP自身已描述得足够好,再加上SAM校正,结合能偏大。
图3可看到,B97系列各种泛函加上SAM色散校正后,除了B972外,一致性地表现为高估色散能,色散作用为主的复合物尤为明显,B973的RMSD为2.05 kJ·mol-1,B97、B971及B97-K的误差在6 kJ·mol-1左右;长程校正ωB97与ωB97X的RMSD达16.65与15.15 kJ·mol-1,个别复合物误差达到-38 kJ·mol-1,这是由于它们自身已有较充分的色散描述,不进行SAM校正时,RMSD值已经达到2.79与2.16 kJ·mol-1,再加上SAM色散能,明显重复,误差较大。
3.2 B972-PFD方法的基函数效应
在确定B972是与SAM色散模型契合度很好的泛函后,初步研究了B972-PFD方法的基组效应,筛选与之相适应的高效率基函数。我们选择了Pople的6-311++G(idjf, kpld)(i = 2, 3, j = 0, 1, k = 1, 2, 3, l = 0, 1)和Dunning相关一致基组cc-pVnZ、aug-cc-pVnZ (n = D, T, Q)以及Ahirichs的def2-TZVP、def2-TZVPP 和def2-QZVP等15种基函数,用S66测试集进行了计算和比较,结合能都使用均衡校正法消除BSSE。
从表2,SI表S4和S5看出,B972-PFD方法计算精度并不随基函数增大而提高;在各种基函数中,6-311++G(2d,p)基是最佳选择,RMSD最小,为0.96 kJ·mol-1,MD、MAD和MAX也最小;同时也发现,当基组不大时,加入弥散函数很有效,6-311G(2d,p)和cc-pVDZ基的RMSD分别为1.30和1.63 kJ·mol-1,加入弥散函数后的6-311++G(2d,p)和aug-cc-pVDZ基,结果明显改善。因此,除一些显著依赖基函数的体系外,我们推荐在消除BSSE的前提下,B972-PFD方法最佳搭配和最优性价比的基函数可选择6-311++G(2d,p)。
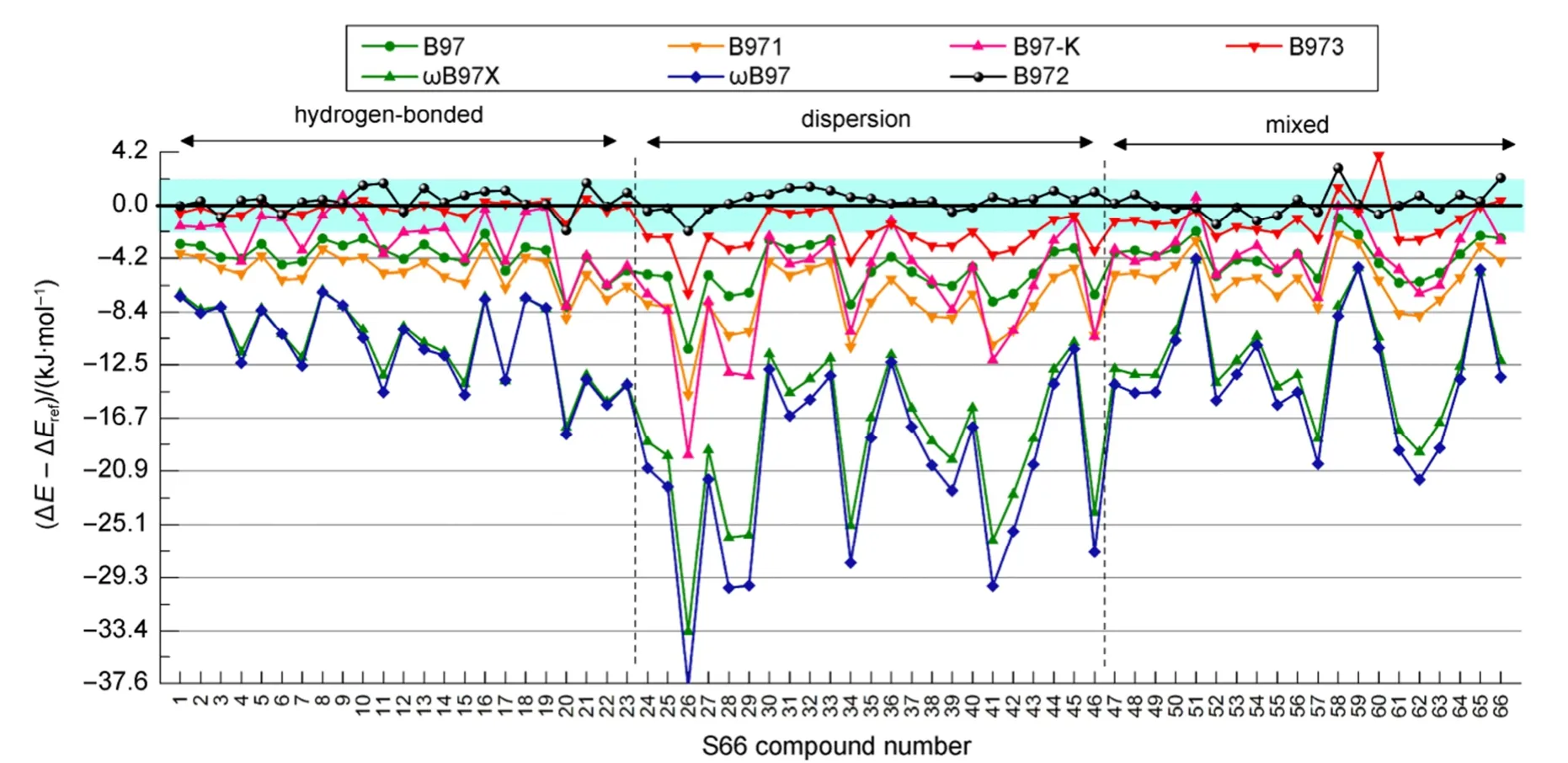
图3 B97系列泛函DFT-PFD/6-311++G(2d,p)以CCSD(T)/CBS为基准的S66数据集误差分析Fig.3 Calculated errors of B97’s DFT-PFD/6-311++G(2d,p) methods with respect to the benchmark CCSD(T)/CBS calculations on the S66 data set
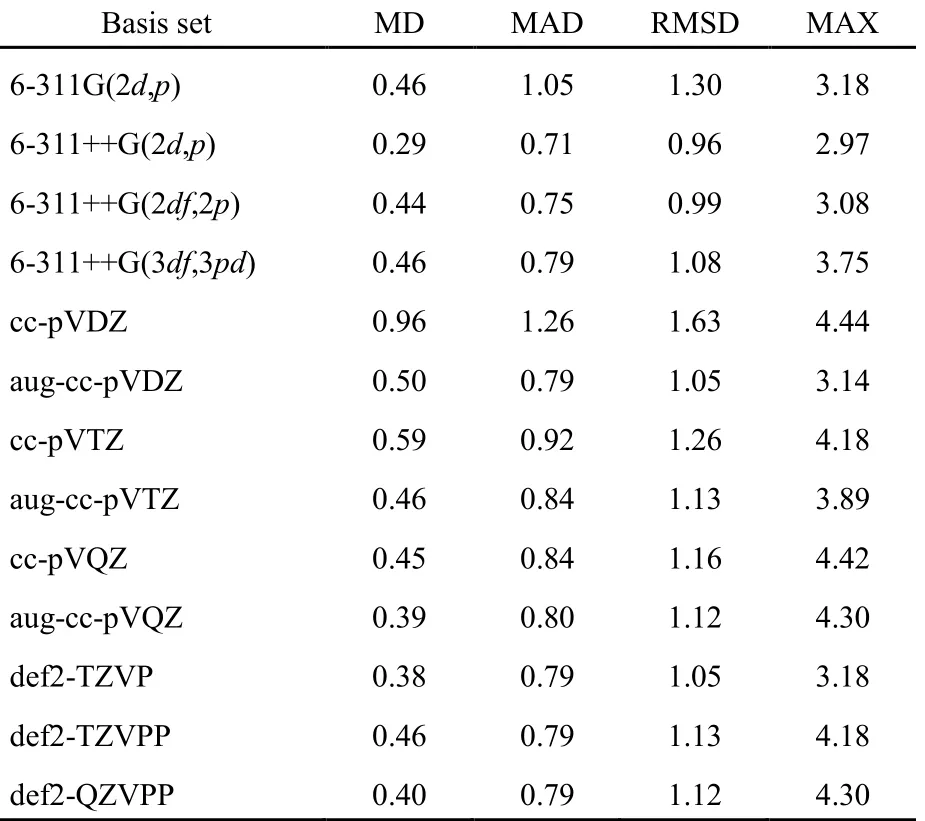
表2 B972-PFD泛函基于S66数据集的基函数效应分析(kJ·mol-1)Table 2 Calculated errors (in kJ·mol-1) of B972-PFD methods for different basis sets based on S66 data set
3.3 B972-PFD方法计算精度分析
3.3.1 S66数据集
我们使用S66数据集,选取了下列四类典型的密度泛函方法与B972-PFD/6-311++G(2d,p)计算结果进行比较测试:首先是B3LYP-D3(BJ)和ωB97X-D44方法,它们近年来被广泛用于分子间相互作用研究,第二是Chai研究组对ωB97X-D改进后的ωB97X-D345泛函,第三是是我们在SAM色散校正原型APF-PFD上改进的APF-PFD*泛函,第四是Head-Gordon研究组近年发展的、迄今为止计算分子间相互作用能最为成功的ωB97X-V、B97M-V和ωB97M-V泛函,最后是双杂化泛函XYG3、XYGJ-OS、ωB97X-2及PBE0-2。测试计算中B3LYP-D3(BJ)使用def2-QZVP基,ωB97X-D和ωB97X-D3用6-311++G(3df,3pd)基,APF-PFD、APF-PFD*、B972-PFD使用6-311++G(2d,p)基,B97M-V、ωB97X-V和ωB97M-V使用aug-cc-pVDZ和aug-cc-pVTZ基,所有的结合能用均衡校正方法消除BSSE,以CCSD(T)/CBS为标准计算误差值。
S66的测试结果见表3和SI表S6及图4,从S66测试集整体来看,APF-PFD*、ωB97X-D和B3LYP-D3(BJ)的RMSD在1.76至1.51 kJ·mol-1之间,处于同一水平,其中APF-PFD*略好于ωB97X-D,稍逊于B3LYP-D3(BJ),而B972-PFD的RMSD为0.96 kJ·mol-1比上述三种方法改进不少;ωB97X-D3的RMSD为1.24 kJ·mol-1比ωB97X-D有较大改进,特别是对色散体系,但逊于B972-PFD;相比ωB97X-V、B97M-V和ωB97M-V/aug-cc-pVDZ(RMSD值0.47、0.93、0.75 kJ·mol-1),aug-cc-pVTZ(RMSD值0.57、0.77、0.53 kJ·mol-1),B972-PFD的RMSD略大,但也小于1 kJ·mol-1;双杂化泛函组XYG3较好,XYGJ-OS和ωB97X-2次之且相近,但都不及B972-PFD。
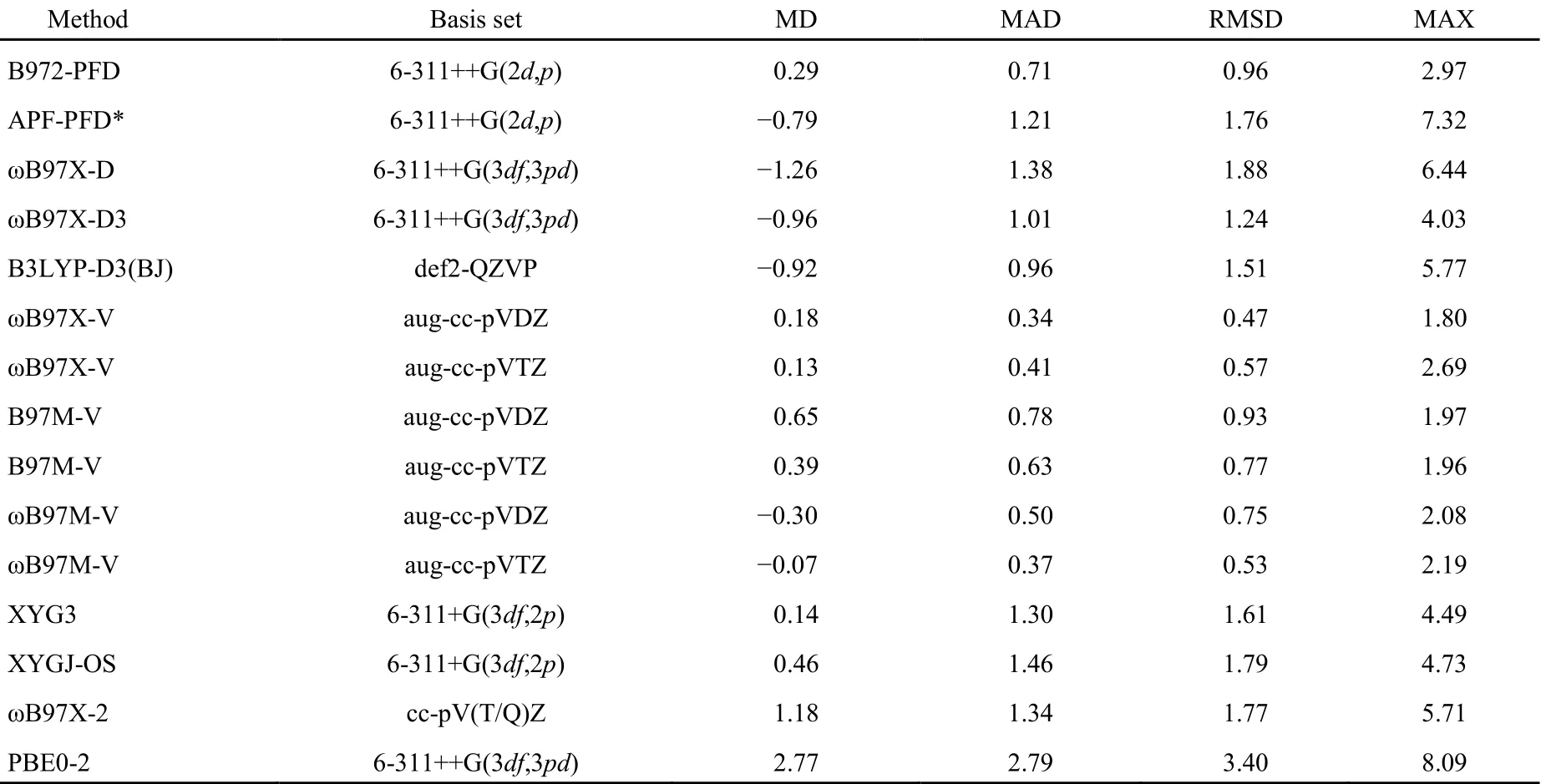
表3 B972-PFD与其它泛函基于S66数据以CCSD(T)/CBS为基准误差分析(kJ·mol-1)Table 3 Calculated errors (in kJ·mol-1) of B972-PFD and other DFT-D methods with respect to the benchmark CCSD(T)/CBS calculations on the S66 data set
从每组复合物单独来看,B972-PFD的最大误差MAX为2.97 kJ·mol-1,比APF-PFD*,ωB97X-D及B3LYP-D3(BJ)小一倍左右,比ωB97X-V、B97M-V和ωB97M-V/aug-cc-pVDZ(MAX值1.80、1.97、2.08),aug-cc-pVTZ(MAX值2.69、1.96、2.19)稍大一些,但仍在化学精度(4.184 kJ·mol-1)范围内。
同时,我们发现B972-PFD误差最大的几组复合物,如以C―H…N作用为主的58号Pyridine二聚体,以N―H…N作用为主66号Methylamine-Pyridine、B97M-V、ωB97X-V和ωB97M-V方法的计算误差也较大,具有一定的相似性,这是以B97泛函为基础的各种衍生方法普遍存在的问题,与此类泛函描述氮原子孤对电子非键作用不足有关,不难看出还有误差稍大的第10、11、21、31、32号复合物中都含氮原子孤对电子。
3.3.2 S22数据集
我们在不同基函数下,测试了S22数据集的B972-PFD与APF-PFD、APF-PFD*、B3LYP-D3(BJ)、B97M-V、ωB97X-V、ωB97M-V等泛函的计算精度,并与XYG3、ωB97X-D和ωB97X-D3文献值列于表4。本文的结合能全部经过BSSE校正。
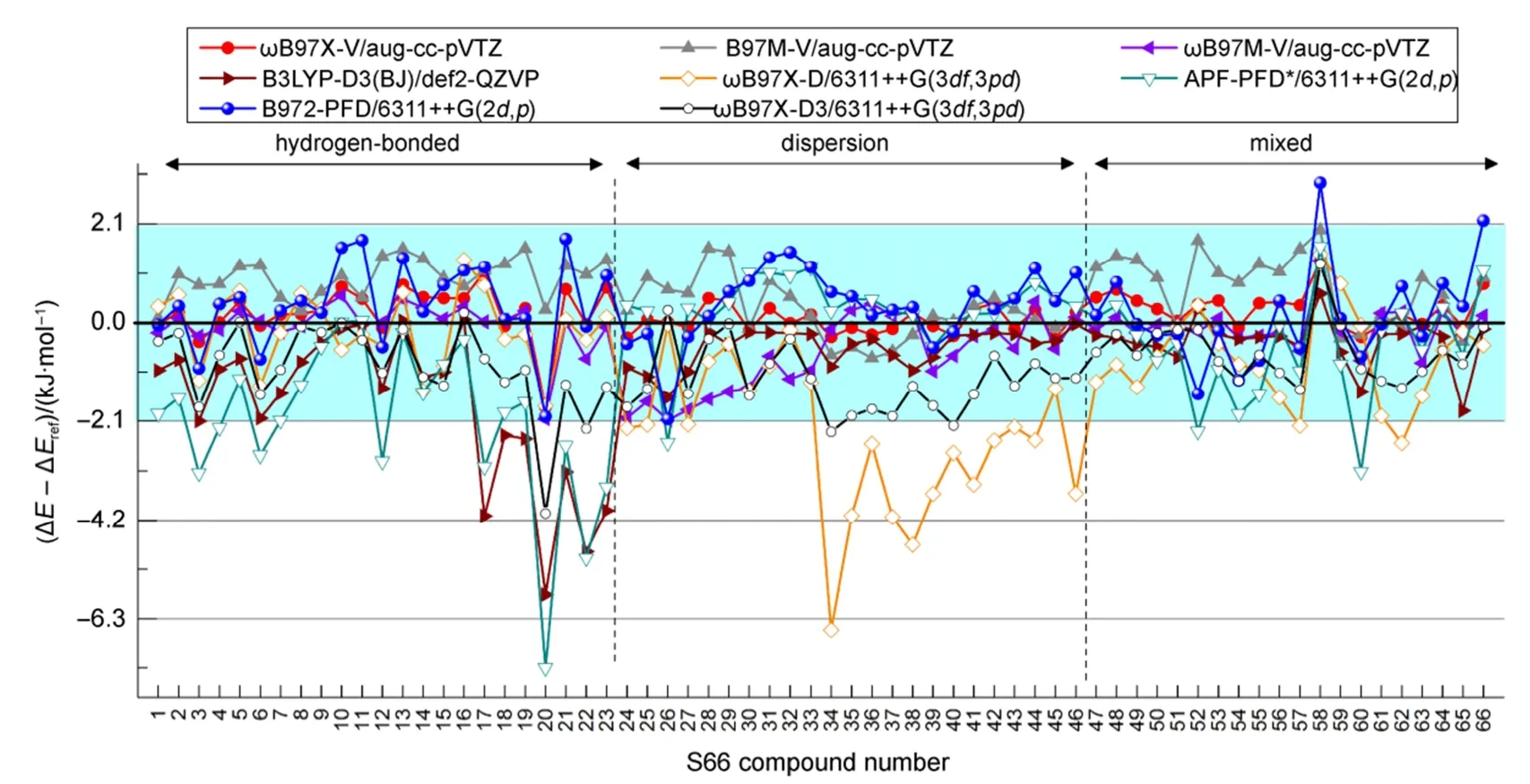
图4 B972-PFD与其他DFT-D泛函方法基于S66数据集的计算精度比较Fig.4 Comparison of calculated precision of B972-PFD and other DFT-D method based on S66 data set
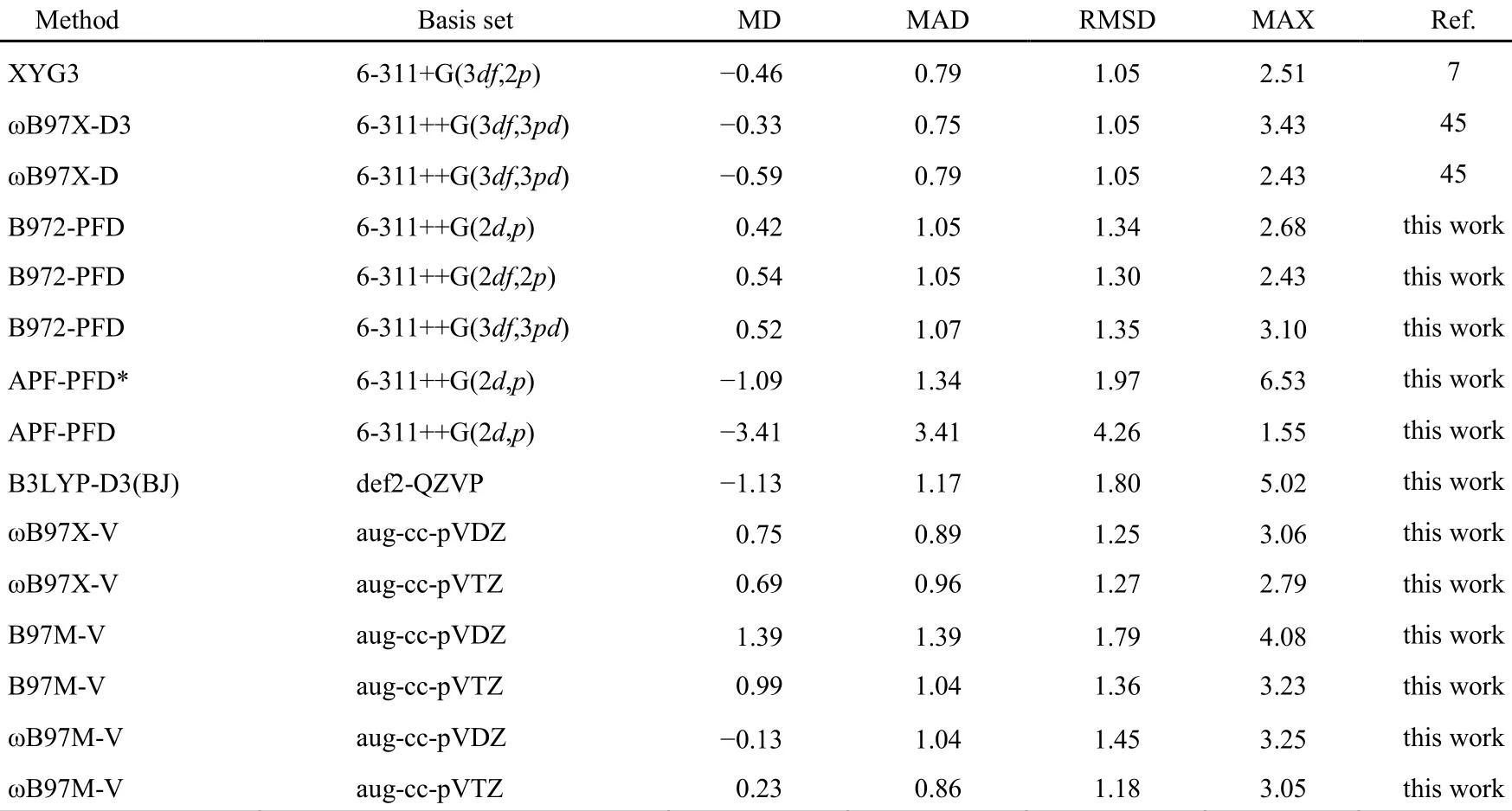
表4 B972-PFD与其它泛函基于S22测试集以CCSD(T)/CBS为基准误差分析(kJ·mol-1)Table 4 Calculated errors (in kJ·mol-1) of B972-PFD and other DFT methods with respect to the benchmark CCSD(T)/CBS calculations on the S22 data set
从表可看到ωB97X-D及其改进版本ωB97X-D3泛函,双杂化泛函XYG3的精度最高,它们的RMSD均为1.05 kJ·mol-1,超过了B972-PFD,甚至超过了在S66数据集测试中表现最佳的ωB97X-V、B97M-V和ωB97M-V;B972-PFD仍然与ωB97X-V、B97M-V和ωB97M-V在相近水平,比B3LYP-D3(BJ)、APF-PFD*及APF-PFD好。加大基函数,对B972-PFD和ωB97X-V计算精度的改善并不明显,而B97M-V和ωB97M-V则很有效。总之,B972-PFD在S22数据集下的表现没有S66那样出色,可能仍然是与B972泛函对氮原子孤对电子非键作用描述不足有关,同样的原因也影响了ωB97X-V、B97M-V和ωB97M-V泛函的计算精度。在此,我们还看到Chai在ωB97X-D3方法中用D3色散校正对此缺陷的修正是成功的45。
3.3.3 大气化学强氢键数据集
对于大气环境化学中硫酸与胺、有机酸伴随质子迁移的强氢键体系,我们测试了B972-PFD与XYG3、M062X、ωB97X-D、ωB97X-V、B97M-V、ωB97M-V/aug-cc-pVTZ的经过BSSE校正后的相互作用能,与CCSD(T)/CBS结果比较于表5。
分析表5的结果,我们发现对于11种强氢键体系的相互作用能,XYG3双杂化泛函表现突出,此类体系正是它的所长之处,B972-PFD与其在同一水平;B972-PFD配合6-311++G(2df,2p)基函数的MD、MAD、MAX及RMSD统计指标明显好于其它5种泛函,也较6-311++G(2d,p)有所改善,增加(f,p)极化函数,有利于强化对体系中硫原子和质子迁移的描述。测试结果预示着B972-PFD在大气气溶胶颗粒形成机制探索中能够发挥作用。
3.3.4 S66x8数据集与非平衡构型下的性能
如前述B972-PFD/6-311++G(2d,p)方法对分子间复合物平衡位置结合能的计算精度较高,但在分子间复合物在非平衡构型下的性能、几何结构优化时性能如何?我们在BSSE校正的前提下,通过对S66x8测试集误差分析和DNA A-T碱基对π…π堆叠和氢键构型势能曲线进行了比较。
S66x8的7个非平衡点和1个平衡位置的误差分析列于表6及SI表S7,我们发现B972-PFD/ 6-311++G(2d,p)方法除了复合物中两单体距离被压缩到平衡距离0.90、0.95倍,处于不稳定排斥状态时,RMSD值稍大外,其它5个非平衡位置的RMSD值均在1 kJ·mol-1水平,与平衡位置的RMSD相近。
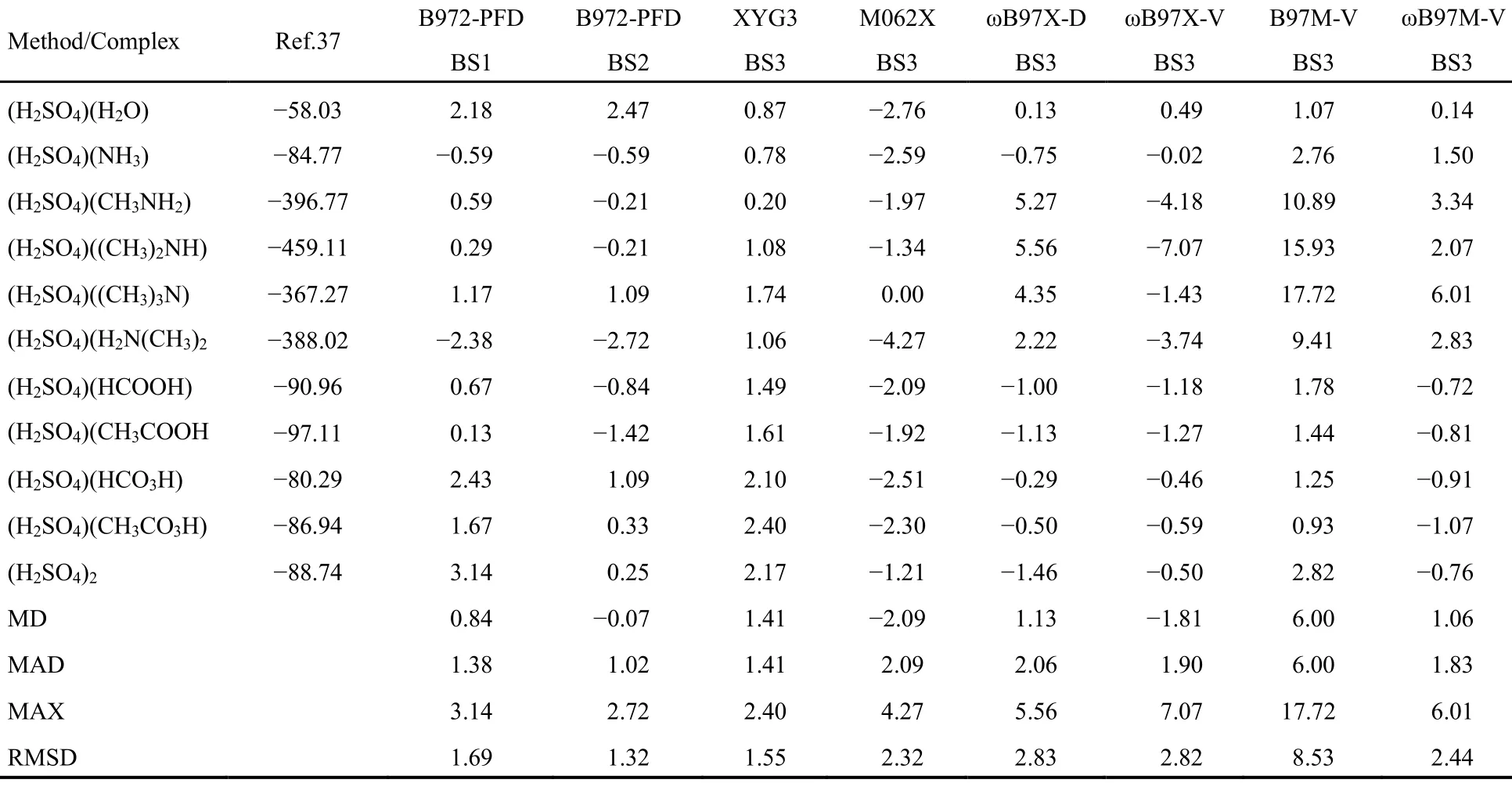
表5 B972-PFD与其它泛函基于大气化学强氢键数据集使用CCSD(T)/CBS基准所得误差分析(kJ·mol-1)Table 5 Calculated errors (in kJ·mol-1) of B972-PFD and other DFT methods with respect to the basis set convergence of the CCSD(T) interaction energies of atmospheric strongly hydrogen-bonded complexes
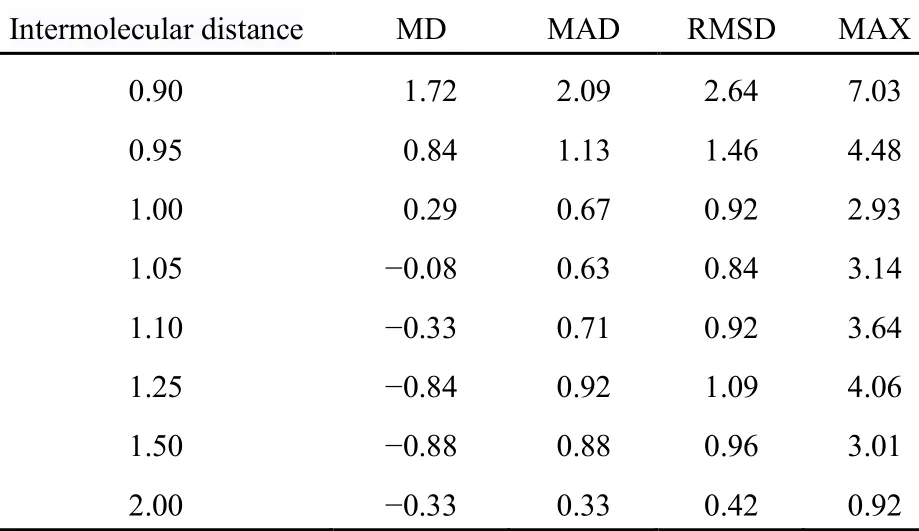
表6 S66x8测试集B972-PFD/6-311++G(2d,p)以CCSD(T)/CBS为基准误差分析(kJ·mol-1)Table 6 Calculated errors (in kJ·mol-1) of B972-PFD/6-311++G(2d,p) with respect to the benchmark CCSD(T)/CBS calculations on the S66x8 data set
最近Martin研究组发表了数十种泛函对S66x8数据集相对于CCSD(T)/CBS的测试结果46,我们选择了其中18种具有代表性泛函与B972-PFD比较于表7。
从表7中,我们发现B972-PFD的总MD为0.04 kJ·mol-1,仅次于B97D3,与TPSS0-D3(BJ)并列排在19种泛函的第二位,它的总RMSD为1.13 kJ·mol-1,排在B3LYP-D3(BJ)、BLYP-D3(BJ)、ωB97X-V之后;从H-bonds、π stacks、London和Mixed分组的RMSD看,B972-PFD在氢键组的表现最佳,在London色散组和混合组次之,在π…π堆叠组表现一般,这可帮助我们把握B972-PFD方法的应用场合。
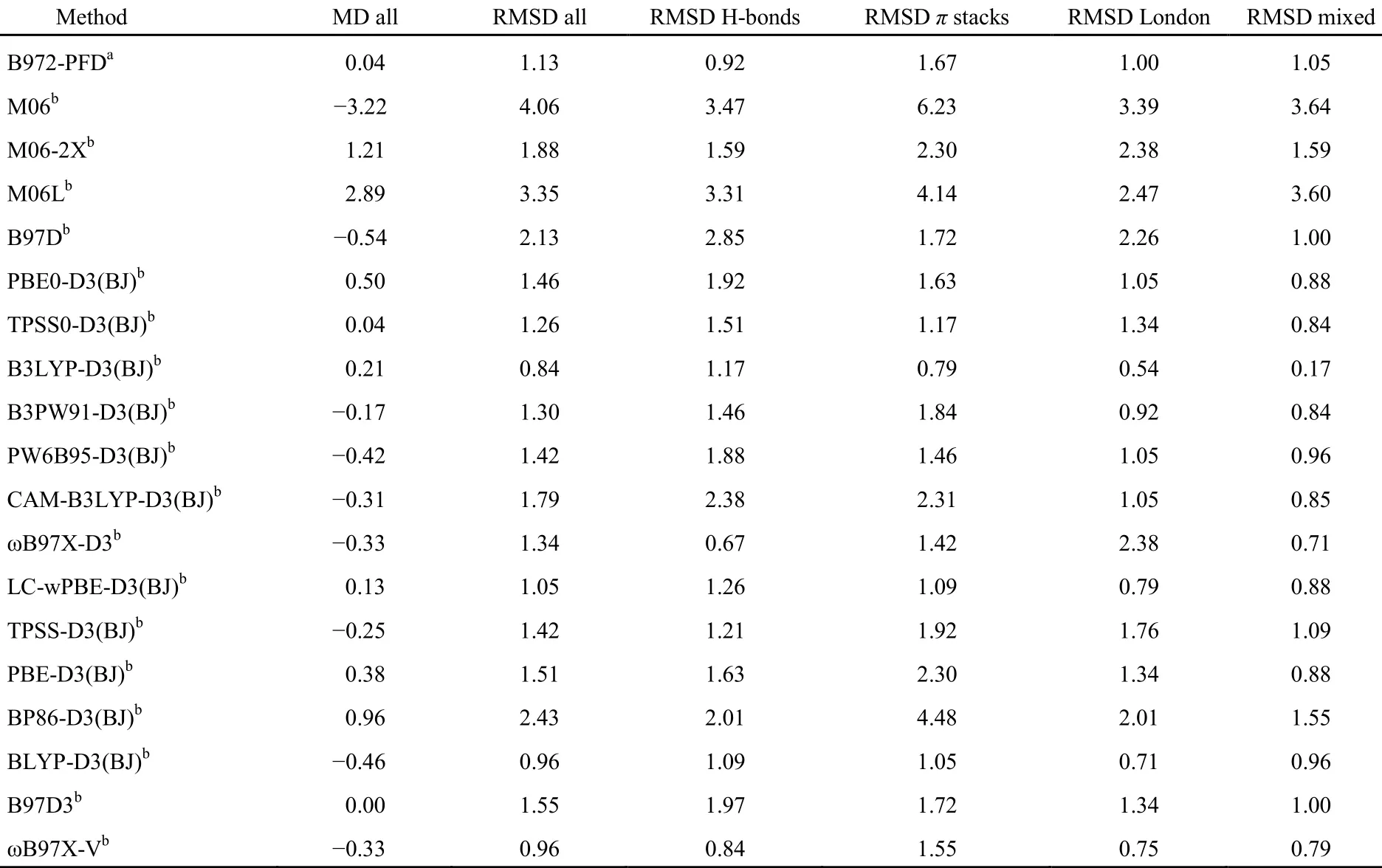
表7 B972-PFD与其它18种DFT-D泛函基于S66x8数据集以CCSD(T)/CBS为基准的误差分析(kJ·mol-1)Table 7 Calculated errors (in kJ·mol-1) of B972-PFD and other DFT-D methods with respect to the benchmark CCSD(T)/CBS calculations on the S66x8 data set
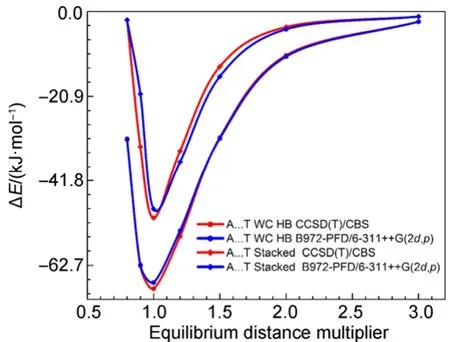
图5 Adenine-Thymine π-π与Adenine-Thymine Watson-Crick复合物在CCSD(T)与B972-PFD方法中的势能曲线Fig.5 Dissociation energy curves of the Adenine-Thymine complex in the π-π stacked and Watson-Crick conformations using CCSD(T) and B972-PFD methods
另外,作为一个经典的例子,我们将A-T DNA碱基对π…π堆叠和氢键两类复合物的B972-PFD/6-311++G(2d,p)与CCSD(T)/CBS势能曲线比较于图5,无论是π…π堆叠还是氢键复合物B972-PFD与CCSD(T)势能曲线重合甚好,极小点位置准确。
两项测试说明在BSSE校正的前提下,B972-PFD/6-311++G(2d,p)方法优化分子间复合物几何结构能够获得较可靠的结果。
3.3.5 CH4与(H2O)20水笼的相互作用
精确计算甲烷与20个水分子构成水笼(H2O)20的相互作用能是可燃冰理论研究中的重要内容,由于甲烷与水分子的平均结合能很小,约为1 kJ·mol-1,还涉及多个水分子的协同效应,计算结果对基函数的极化和弥散部分也较敏感,是挑战各种理论方法的课题。前人已用QMC、MP2、MP2C、att-MP2、ωB97X-V、ωB97X-D、LC-VV10、 M06-2X、M06-2X-D3、B2PLYP、B2PLYP-D3、DFT-SAPT、XSAPT(KS)+D3等方法进行过计算,CH4与(H2O)20作用能的计算值从-4.56到-26.71 kJ·mol-1,差别较大47,48。由于还没有CCSD(T)/CBS结果发表,量子Monte Carlo (QMC)方法获得的
a6311++G(2d,p);bdef2-QZVP, Ref.46(-22.15 ± 2.09) kJ·mol-1数值是目前最可信的参考标准。我们分别用6-311++G(2d,p)、aug-cc-pVTZ和aug-cc-pVQZ基组,对B972-PFD方法进行了测试计算,同时还增加了APF-PFD、APF-PFD*、B3LYP-D3(BJ)、B97M-V、ωB97M-V、ωB97X-D3、XYG3和XYGJ-OS的对照计算,各种方法的结果比较于表8。
从表8我们可看到B972-PFD泛函表现出色,与APF-PFD*、B97M-V和ωB97M-V三种泛函的计算结果相近,同时也与XYG3和XYGJ-OS双杂化泛函结果差别不大。但对于此类基函数依赖性很强的体系,使用比6-311++G(2d,p)更大的基组,如aug-cc-pVTZ及aug-cc-pVQZ能获得更好的结果。

表8 CH4与(H2O)20作用能计算(kJ·mol-1) Table 8 Binding energy (in kJ·mol-1) of CH4to (H2O)20
4 结 论
SAM色散校正模型具备物理图像清晰、简单明了,是一类值得关注的色散校正方法,其应用的关键是找到与之契合的密度泛函。我们在常见泛函中筛选出的B972是与SAM色散校正模型契合度最好的密度泛函;通过基函数效应研究,发现在大多数场合,6-311++G(2d,p)基组是B972-PFD方法的最优组合。
计算测试表明,B972-PFD/6-311++G(2d,p)是一种计算量仅在杂化泛函水平、高精度的DFT-D方法,对于不同尺寸分子间的氢键、色散及混合作用均有较好的描述,同时具备可靠的几何结构优化特性。并且,若采用Gaussian 09 Rev.D01/E01或Gaussian 16程序,B972-PFD方法无需修改程序或挂接附加程序,用“B972 Empirical Dispersion = PFD”关键字,即可直接计算能量,还能方便地计算能量的一阶与二阶解析梯度,高效地优化体系几何结构、计算谐振频率及热力学函数等,具有良好的应用前景。
Supporting Information: available free of charge via the internet at http://www.whxb.pku.edu.cn.
(1) Rezac, J.; Riley, K. E.; Hobza, P. J. Chem. Theory Comput. 2011, 7, 2427. doi: 10.1021/ct200294
(2) Yu, H. S.; He, X.; Li, S. L.; Truhlar, D. G. Chem. Sci. 2016, 7, 5032, doi: 10.1039/C6SC00705H
(3) Mardirossian, N.; Head-Gordon, M. J. Chem. Theory Comput. 2016, 12, 4303. doi: 10.1021/acs.jctc.6b00637
(4) Schwabe, T.; Grimme, S. Phys. Chem. Chem. Phys. 2007, 9, 3397. doi: 10.1039/B704725H
(5) Chai, J. D.; Mao, S. P. Chem. Phys. Lett. 2012, 538, 121. doi: 10.1016/j.cplett.2012.04.045
(6) Chai, J. D.; Head-Gordon, M. J. Chem. Phys. 2009, 131, 174105. doi: 10.1063/1.3244209.
(7) Zhang, Y.; Xu, X.; Goddard, W. A., III. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 4963. doi: 10.1073/pnas.0901093106
(8) Zhang, I. Y.; Xu, X.; Jung, Y.; Goddard, W. A., III. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 19896. doi:10.1073/pnas.1115123108
(9) Grimme, S.; Hansen, A.; Brandenburg, J. G.; Bannwarth, C. Chem. Rev. 2016, 116 (SI), 5105. doi: 10.1021/acs.chemrev.5b00533.
(10) Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. J. Chem. Phys. 2010, 132, 154104. doi: 10.1063/1.3382344
(11) Vydrov, O. A.; Van Voorhis, T. J. Chem. Phys. 2010, 133, 244103. doi: 10.1063/1.3521275
(12) Mardirossian, N.; Head-Gordon, M. Phys. Chem. Chem. Phys. 2014, 16, 9904. doi: 10.1039/c3cp54374a
(13) Mardirossian, N.; Head-Gordon, M. J. Chem. Phys. 2015, 142, 074111. doi: 10.1063/1.4907719
(14) Mardirossian, N.; Head-Gordon, M. J. Chem. Phys. 2016, 144, 214110. doi: 10.1063/1.4952647
(15) Austin, A.; Petersson, G. A.; Frisch, M. J.; Dobek, F. J.; Scalmani, G.; Throssell, K. J. Chem. Theory Comput. 2012, 8, 4989. doi: 10.1021/ct300778e
(16) Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; et al. Gaussian 09, Revision D.01; Gaussian Inc.: Wallingford CT, 2009.
(17) Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; et al. Gaussian 16, Rev. A.03; Gaussian Inc.: Wallingford CT, 2016.
(18) He, Y.; Wang, Y. B. Acta Phys. -Chim. Sin. 2016, 32, 2709. [何 禹,王一波. 物理化学学报, 2016, 32, 2709] doi: 10.3866/PKU.WHXB201609132
(19) Wilson, P. J.; Bradley, T. J.; Tozer, D. J. J. Chem. Phys. 2001, 115, 9233. doi: 10.1063/1.1412605
(20) Becke, A. D. J. Chem. Phys. 1993, 98, 5648. doi: 10.1063/1.464913
(21) Becke, A. D. Phys. Rev. A 1988, 38, 3098. doi: 10.1103/PhysRevA.38.3098
(22) Perdew, J. P.; Burke, K.; Ernzerhof, M. Phys. Rev. Lett. 1996, 77, 3865. doi: 10.1103/PhysRevLett.77.3865
(23) Adamo, C.; Barone, V. J. Chem. Phys. 1999, 110, 6158. doi: 10.1063/1.478522
(24) Becke, A. D. J. Chem. Phys. 1996, 104, 1040. doi: 10.1063/1.470829
(25) Tao, J. M.; Perdew, J. P.; Staroverov, V. N.; Scuseria, G. E. Phys. Rev. Lett. 2003, 91,146401. doi: 10.1103/PhysRevLett.91.146401
(26) Chai, J. D.; Head-Gordon, M. J. Chem. Phys. 2008, 128, 084106. doi: 10.1063/1.2834918
(28) Hamprecht, F. A.; Cohen, A.; Tozer, D. J.; Handy, N. C. J. Chem. Phys. 1998, 109, 6264. doi: 10.1063/1.477267
(29) Thomas, W. K.; David, J. T. J. Chem. Phys. 2005, 123, 121103. doi: 10.1063/1.2061227
(30) Boese, A. D.; Martin, J. M. J. Chem. Phys. 2004, 121, 3405. doi: 10.1063/1.1774975
(31) Xu, X.; Goddard, W. A., III. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 2673. doi: 10.1073/pnas.0308730100
(32) Dunning, T. H. J. Chem. Phys.1989, 90, 1007. doi: 10.1063/1.456153
(33) Kendall, R. A.; Dunning, T. H.; Harrison, R. J. J. Chem. Phys.1992, 96, 6796. doi: 10.1063/1.462472
(34) Woon, D. E.; Dunning, T. H. J. Chem. Phys. 1993, 98, 1358. doi: 10.1063/1.464303
(35) Weigend, F.; Ahlrichs, R. Phys. Chem. Chem. Phys. 2005, 7, 3297. doi: 10.1039/b508541a
(36) Jurecak, P.; Sponer, J.; Cemy, J.; Hobza, P. Phys. Chem. Chem. Phys. 2006, 8, 1985. doi:10.1039/b600027d
(37) Elm, J.; Kristensen, K. Phys. Chem. Chem. Phys. 2017, 19, 1122. doi:10.1039/c6cp06851k
(38) Rezac, J.; Riley, K. E.; Hobza, P. J. Chem. Theory Comput. 2011, 7, 3466. doi: 10.1021/ct200523a
(39) Shao, Y.; Gan, Z.; Epifanovsky, E.; Gilbert, A. T.; Gill, P. M.; Head-Gordon, M. Mol. Phys. 2015, 113, 184. doi: 10.1080/00268976.2014.952696
(40) Wang, Y. B.; Lin, Z. J. Am. Chem. Soc. 2003, 125, 6072. doi: 10.1021/ja028998g
(41) Wang, W. Z.; Sun, T.; Zhang, Y.; Wang, Y. B. J. Chem. Phys. 2015, 143, 4145. doi: 10.1063/1.4931121
(42) Sun, T.; Wang Y. B. Acta Phys. -Chim. Sin. 2011, 27, 2553. [孙 涛,王一波. 物理化学学报, 2011, 27, 2553.] doi: 10.3866/PKU.WHXB20111017
(43) Li, M. M.; Wang, Y. B.; Zhang, Y.; Wang, W. Z. J. Phys. Chem. A 2016, 120, 5766. doi: 10.1021/acs.jpca.6b06492
(44) Chai, J. D.; Head-Gordon, M. Phys. Chem. Chem. Phys. 2008, 10, 6615. doi: 10.1039/b810189b
(45) Lin, Y. S.; Li, G. D.; Mao, S. P.; Chai, J. D. J. Chem. Theory Comput. 2013, 9.263. doi:10.1021/ct300715s
(46) Brauer, B.; Kesharwani, M. K.; Kozuch, S.; Martin, J. M. Phys. Chem. Chem. Phys. 2016, 18, 20905. doi: 10.1039/c6cp00688d.
(47) Deible, M. J.; Tuguldur, O.; Jordan K. D. J. Phys. Chem. B 2014, 118, 8257. doi: 10.1021/jp501592h
(48) Lao, K. U.; Herbert, J. M. J. Phys. Chem. A 2015, 119, 235. doi: 10.1021/jp5098603
B972-PFD: A High Accuracy Density Functional Method for Dispersion Correction
HE Yu1,2WANG Yi-Bo1,2,*
(1Key Laboratory of High Performance Computational Chemistry, Guiyang 550025, P. R. China;2Network and Information Center of Guizhou University, Guiyang 550025, P. R. China)
A novel DFT-D method, B972-PFD, has been found by combining the B972 hybrid density functional with the empirical dispersion correction based on the spherical atom model (SAM). The performance of the B972-PFD method is assessed on the S66, S66x8, and S22 standard data sets, atmospheric hydrogen-bonded clusters, the Adenine-Thymine π…π stacked, Watson-Crick hydrogen-bonded complexes, and the methane to (H2O)20water cluster. The benchmark results of the S66 test set show that B972-PFD and three recently developed density functionals, ωB97X-V, B97M-V, and ωB97M-V developed by the Head-Gordon group, are at the same level of accuracy, and have an root-mean-square deviation (RMSD) of binding energies less than 1 kJ·mol-1relative to the CCSD(T)/CBS gold standard. The B972-PFD method also showed excellent accuracy in other data set tests. The basis set effect of the B972-PFD method has been benchmarked, and we recommend that the favorable price/performance ratios basis set is Pople΄s 6-311++G(2d,p).
Intermolecular Interaction; DFT-D; Spherical atom model for dispersion correction; B972-PFD
January 10, 2017; Revised: March 9, 2017; Published online: March 29, 2017.
O641
Becke, A. D. J. Chem. Phys. 1997, 8554.
10.1063/1.475007
doi: 10.3866/PKU.WHXB201703291
*Corresponding author. Email: ybw@gzu.edu.cn; Tel: +86-851-88292009.
The project was supported by the National Natural Science Foundation of China (41165007) and Natural Science Foundation of Guizhou Province, China (20082116).
国家自然科学基金(41165007)和贵州省自然科学基金(20082116)资助项目
© Editorial office of Acta Physico-Chimica Sinica
猜你喜欢
中学生数理化·八年级物理人教版(2022年10期)2022-11-10
高中数理化(2022年14期)2022-08-15
中学生数理化·八年级物理人教版(2021年10期)2021-11-22
中学生数理化·八年级物理人教版(2020年11期)2020-12-14
中学生数理化·八年级物理人教版(2019年10期)2019-11-25
原子与分子物理学报(2019年5期)2019-04-28
广东造船(2018年1期)2018-03-19
中学化学(2015年12期)2016-01-19
建筑科学与工程学报(2014年1期)2014-08-08
原子与分子物理学报(2014年3期)2014-02-28
