
双链阴离子表面活性剂1-烷基-癸基磺酸钠在气/液界面聚集行为:分子动力学模拟研究
2017-06-21 12:33陈贻建周洪涛葛际江徐桂英
物理化学学报 2017年6期
陈贻建 周洪涛 葛际江 徐桂英
(1中国石油大学(华东)石油工程学院,山东 青岛266580;2山东大学胶体与界面化学教育部重点实验室,济南 250100)
双链阴离子表面活性剂1-烷基-癸基磺酸钠在气/液界面聚集行为:分子动力学模拟研究
陈贻建1,*周洪涛1葛际江1徐桂英2
(1中国石油大学(华东)石油工程学院,山东 青岛266580;2山东大学胶体与界面化学教育部重点实验室,济南 250100)
采用分子动力学模拟方法研究了双链阴离子表面活性剂1-烷基-癸基磺酸钠(1-Cm-C9-SO3Na)在气/液界面的聚集行为。通过分析体系中各组分的密度分布和径向分布函数,考察了m大小对其界面性质的影响。结果表明:随着m的增大,表面活性剂的疏水性增强,疏水碳链的倾斜角也随之降低;m = 4时,1-C4-C9-SO3Na分子采用平躺的方式在界面上聚集,S-Na+和S-S的相互作用最大,极性头基的水化能力最弱。通过模拟和实验对比得出,m增加到4个时,对该类双链阴离子表面活性剂性能的提高最显著。
分子动力学模拟,1-烷基-癸基磺酸钠(1-Cm-C9-SO3Na);气-液界面,聚集行为
1 引 言
烷基磺酸盐是被广泛应用的阴离子型表面活性剂,尤其是强化采油技术的发展,促使人们对磺酸盐表面活性剂的研究兴趣越来越浓, 近年来,烷基磺酸盐类阴离子型表面活性剂仍然是研究者感兴趣的课题1-6。李之平等7曾对烷基磺酸盐中烷基链长度对相行为的影响进行了研究,发现最佳盐浓度(刚出现三相和三相刚消失时水相中的盐浓度平均值)的对数值与烷基磺酸钠中碳原子数有线性关系,长链的具有较低的最佳盐浓度。肖进新等8发现尽管烷基磺酸盐与烷基硫酸盐在分子结构上仅仅在头基上相差一个氧原子,但其水溶性大不相同。分子内含有一个亲水基团和两个憎水基团的双烃链表面活性剂相对于由单疏水链和单极性基团组成的表面活性剂具有许多独特之处9。例如,二(2-乙基己基)琥珀酸酯磺酸钠(AOT)在非极性溶剂中极易形成球形聚集体,且聚集体尺寸随体系含水量增大而大10。于涛等11的研究也表明,十四烷基芳基磺酸盐分子亲油基支化度的增加,不利于其在极性溶剂中形成胶束,而易在非极性溶剂中形成反相胶束。他们还发现12,13,随芳基向烷基链中间位置移动,分子的支化程度增加,疏水基团的空间体积增大,会导致其在油砂上的吸附量降低。烷基链长度的增加,或者芳基向烷基链中间位置移动,其所形成的微乳液体系增溶能力增强,但耐盐能力下降。我们曾研究过AOT和4-(5'-十六烷基)苯磺酸钠(5C16)在界面的聚集行为。结果表明,AOT在气/液界面上的排列是粗糙不平的,其碳氢链尾巴呈弯曲状朝向气相14;而5C16分子的两支链会形成差距30°的两个角度分布,当CaCl2存在时,受静电势的影响聚集结构更加紧密,两个角度分布所占比例从 1.9 : 1变为4.0 : 115。Griffin等16也发现Ca2+可以使AOT在云母/水界面上形成致密的多层结构。
显然,表面活性剂的结构对其聚集行为有着显著的影响。为了解释表面活性剂的聚集行为及其微观作用机理,人们对计算机模拟在两亲分子研究中的应用愈来愈重视17-22。分子动力学模拟可以从微观上揭示表面活性剂结构与其性能之间的关系。曹绪龙等23的模拟结果表明,温度升高会使得烷基α-烯烃磺酸盐的疏水链变得弯曲,且相互缠绕交错,从而使其形成的界面膜强度增大,所以超长链(碳数多于20个)的烷基α-烯烃磺酸盐形成的泡沫在高温下有较好的稳定性。丁伟等24通过模拟发现:随着分子结构中芳环向碳链中间位置移动,烷基芳基磺酸盐的胶束化能力和胶束稳定性均下降。Liu等25模拟了油水界面上支化的烷基苯磺酸盐表面活性剂的聚集行为,发现疏水部分尺寸越大,使得其空间位阻越大,从而会降低界面上表面活性剂相变速度。He等26也模拟了一系列烷基苯磺酸在气-液界面的单层结构,发现体系的表面张力和烷基苯磺酸分子的倾斜角与烷基链的长度和支化密切相关,而烷基苯磺酸的溶解性和迁移能力则主要有烷基链长度决定。Kabra等27对比研究正丁基苯磺酸钠、异丁基苯磺酸钠和叔丁基苯磺酸钠在气-液界面的聚集行为时发现,丁基的几何构型对其分子的界面占有面积有显著的影响,说明碳链的支化度对表面活性剂的性能有着重要的影响。由此可见,通过分子动力学模拟方法可以探讨表面活性剂微观结构与宏观性能之间的构效关系。这对新型表面活性剂的设计合成以及性能表征都有重要的意义。在分子动力学模拟方法研究表面活性剂聚集行为方面,本课题组已进行过许多研究28-33。本文主要利用分子动力学模拟的方法研究了1-烷基-癸基磺酸钠(1-Cm-C9-SO3Na)系列表面活性剂在气-液界面的聚集行为,从微观角度探讨m的改变对表面活性剂界面聚集行为的影响,以期为该类表面活性剂的应用以及未来新型烷基磺酸类表面活性剂的设计合成提供理论指导。
2 模拟方法
图1给出了1-烷基-癸基磺酸钠(1-Cm-C9-SO3Na)的化学结构式以及在模拟中各原子上的电荷分布情况。为了简化需要,氢原子的电荷未给出。根据实验上得到的1-烷基-癸基磺酸钠饱和吸附时的最小分子截面积在0.57到1.04 nm234,再根据我们模拟的格子尺寸得到水表面面积为3 nm × 3 nm = 9 nm2,从而使得模拟体系中单个表面活性剂的分子截面积为1.6 nm2,确保所有模拟体系中,表面活性剂浓度在水面上都处在饱和吸之前的状态。利用Amorphous Cell模块构建水分子,在水分子层上下两面各安放5个均匀排列在水表面,并使每个垂直于x、y轴组成的平面,从而使得模拟体系中单个表面活性剂的分子截面积为1.6 nm2。把的极性头部分插入含1000个水分子的厚度为3.3 nm的水层中,水分子采用SPC势能模型。3.3 nm厚度的水层是为了保证水层中的水分子性质接近本体溶液, 同时保证水层上下的两个单层独立,不会相互影响。紧接着随机向水盒子中加入从1-Cm-C9-SO3Na分子中分离出来的等数量的Na+离子,确保体系的电中性。为消除z方向上的周期性重复对体系的影响,在体系两侧分别加足够厚度的真空层(大于5nm),模拟体系的格子尺寸为3 nm × 3 nm × 20 nm。
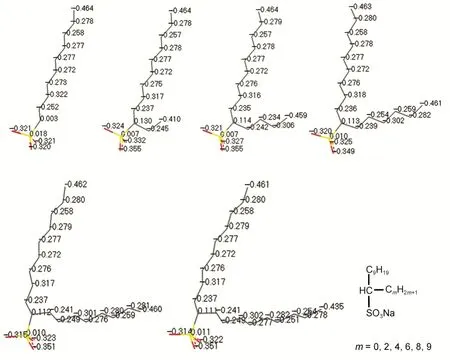
图1 1-Cm-C9-SO3Na的化学结构示意图及各原子上的电荷Fig.1 Scheme of chemical structures for 1-Cm-C9-SO3Na and charge distribution in molecules Hydrogen atoms are not shown.
所有模拟均在Material Studio软件中运行,采用NVT系综,引入周期边界条件,选取COMPASS力场,该力场的非键相互作用能通过公式(1)计算。使用Hoover-Nóse控温方法,温度控制在298K,弛豫时间为0.2 ps。模拟时间为2 ns,步长为1 fs,最后1 ns存储的轨迹文件用于后续结构和动力学分析。长程静电势采用Ewald方法,截断半径设定为0.95 nm。模拟体系构型建立之后,先用Smart方法进行能量最小化,然后再开始进行动力学模拟。
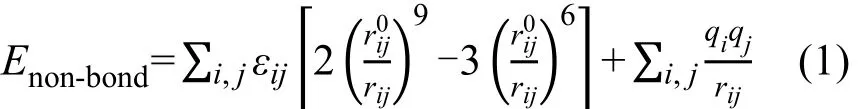
式中rij、和εij分别为粒子i和j之间的距离、尺度和能量参数,qi和qj分别为粒子i和j的电荷。
3 结果和讨论
3.1 表面活性剂界面聚集行为
所有体系表面活性剂均在界面聚集,如图2所示。随着m的增加,表面活性剂进入界面层水相的程度逐渐减弱。由图2A中明显看出,整个表面活性剂分子几乎都聚集在界面层中,而随着m的增加,表面活性剂的部分碳氢链逐渐远离界面区域 (图2B、2C和2D),最终绝大部分碳氢链都呈现远离界面水层的趋势(图2E和2F)。这表明,随着m的增加,1-Cm-C9-SO3Na的疏水性增强,碳氢链逐渐远离界面水相区域,而更多地进入气相。从碳链的排列来看,在m = 0,4时(图2A和2C),碳链基本以平躺为主,而其它几个体系,碳链则以倾斜状居多。Na+则多集中在界面区域,部分会留在水相当中。
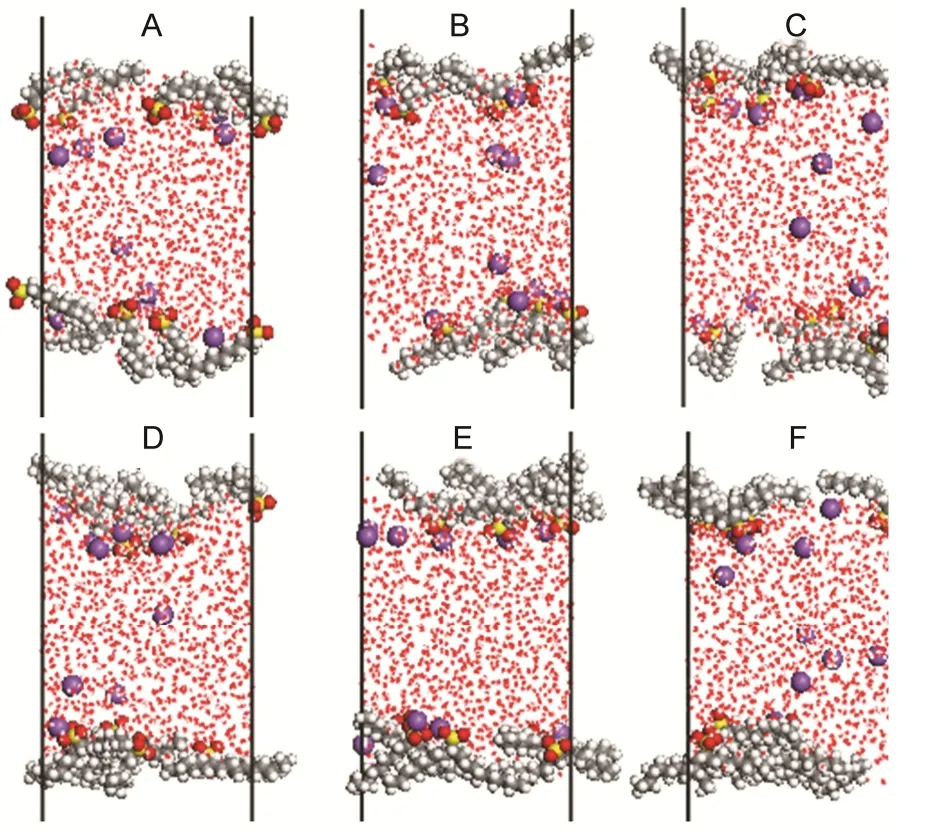
图2 各体系模拟平衡时的构型Fig.2 Balanced configurations for all systems
更为详细的结构特征可以通过解析体系中各组分的密度分布函数获得。图3给出了各体系中H2O、和Na+的密度分布函数图。从的分布图可以看出,所有体系的峰均较尖,峰也基本位于界面区域,说明在气-液界面形成有序的单层。Na+的分布较为广泛,水相也有小峰,界面区域有最高峰,表明Na+主要分布在界面区域,这与图2中直观看到的结果一致。为了更直观比较m的增加对和Na+密度分布的影响,将密度分布图中两个组分的最高峰出峰位置以及峰高列于表1当中。从的出峰位置可以看出,m = 0时,其峰位置和峰值均要小于其它体系,说明随着m的增加,表面活性剂的疏水性增强,其分布必然要呈现远离水相的趋势,故而出峰位置更大,而疏水性增强有利于其在界面形成聚集体,所以密度较高(峰值大)。从图3A中也可以发现,的峰有更大一个区域与水相重叠,说明其亲水性更强。且出峰区域大约是从1.0到2.5 nm,要宽于其它体系(1.2 到2.5 nm),导致其峰值较低。从Na+的峰位置及峰高可以看到,m = 0时,其峰位置和峰值也大体小于其它体系,这应该是由表面活性剂的头基引起的,从图1的电荷分布结果可以得到m = 2,4,6,8,9时,表面活性剂头基的带电量分别为-1.004e、-0.996e、-0.984e、-0.979e和-0.976e,随着m的增加,负电性逐渐降低,但含有侧链的头基负电性均大于m = 0的-0.943e,吸引更多的Na+进入界面区域,所以峰值增大,而随着m增加,表面活性剂是远离水相的,必然引起Na+向界面区域移动,故而出峰位置增大。随着m的增加,和Na+最高峰的位置及峰高的变化规律并未呈现单一的线性关系,这可能是由于m的增加,头基负电量增大对Na+静电吸

图3 各组分密度分布曲线Fig.3 Density profiles of different components
表1 密度分布图中和Na+最高峰的位置及峰高Table 1 Position and values of the highest peak ofand Na+in the density profiles
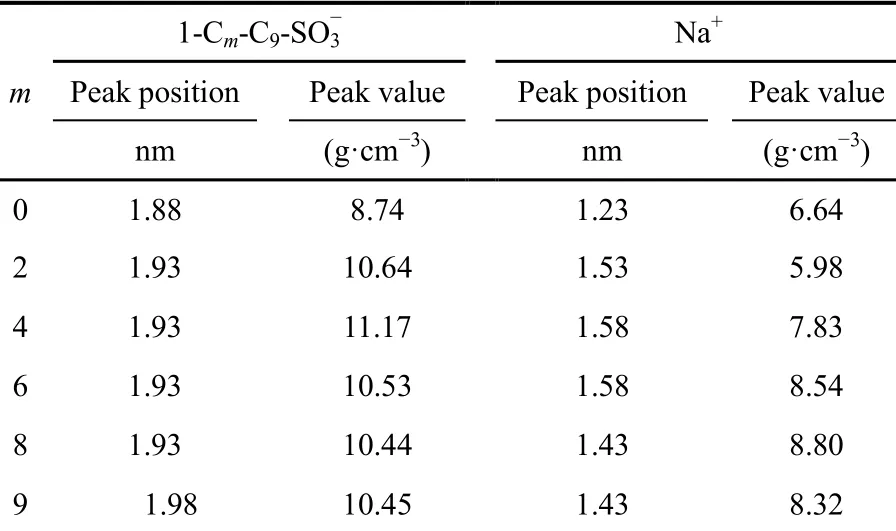
表1 密度分布图中和Na+最高峰的位置及峰高Table 1 Position and values of the highest peak ofand Na+in the density profiles
1-Cm-C9-SO-3 Na+m Peak position Peak value Peak position Peak value nm (g·cm-3) nm (g·cm-3) 0 1.88 8.74 1.23 6.64 2 1.93 10.64 1.53 5.98 4 1.93 11.17 1.58 7.83 6 1.93 10.53 1.58 8.54 8 1.93 10.44 1.43 8.80 9 1.98 10.45 1.43 8.32
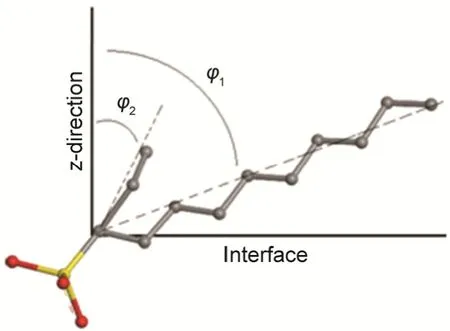
图4 碳链在界面上倾斜角的示意图Fig. 4 Scheme of slant angle of carbon chains at the interface
众所周知,表面活性剂分子在界面上并非垂直排列的,为进一步考察其吸附行为,通过以下公式计算了表面活性剂分子在气-液界面上的倾斜角(φ)。

式中l为碳链的长度,lz为其在界面法线方向上的投影长度,倾斜角(φ)为表面活性剂分子与界面法线之间的角度。图4给出了倾斜角的示意图。通过上述公式,求得1-Cm-C9-SO3Na分子中碳链的倾斜角,列于表2中。长链指的是9个碳的碳氢链,短链则是另一条碳氢链,当然当m = 0时,只有一个长链,当m = 9时,两个链都可以是长链,为了对比结果,同样分成了两个链。各碳链的结构(理论)长度分别为l(C2) = 0.26、l(C4) = 0.52、l(C6) = 0.76、l(C8) = 1.01、l(C9) = 1.14 nm。从表2可以看出,长链的l值小于其结构长度,表明长链在气-液界面并非是完全伸直的。随着m的增加,l逐渐增大,说明长链逐渐拉伸,这主要是由于m增大,即碳原子数增多,占据的面积就更大。在界面区域面积固定的情况下,长链需采取更为节省空间的方式,碳链会逐渐从弯曲状向拉直状转变,l逐渐增加。φ1的结果也表明,m = 0时,其倾斜角度最大,长碳链更靠向界面区域,这从图2A中可以发现,此时的碳链基本是平躺在水面上。
m增加后,其倾斜角明显降低,长链逐渐远离界面,往法线(垂直于界面)方向靠近。这主要是m增加后,表面活性剂碳氢链的疏水性增强,逐渐远离界面,而极性头基依然位于水相。故而表面活性剂在气-液界面采取更为直立的方式,所以φ1减小。该结论与的密度分布结果基本一致。从表2中短链的倾斜角φ2可以发现,m = 4时,倾斜角为82.3o远远大于其它体系,且非常接近90o,表明此时的短尾链基本与水面平行,平躺在界面上。从φ1的结果也发现,所有双链体系中,m = 4时的倾斜角也是最大的。这说明当m = 4时,表面活性剂分子采取最为平躺的方式吸附在界面上,单个分子所占据的界面面积最大,所以少量分子就可达到吸附饱和状态,其临界聚集浓度(cmc)降低幅度增大。

表2 模拟平衡时碳链长度及其倾斜角Table 2 Balanced length and slant angles of carbon chains
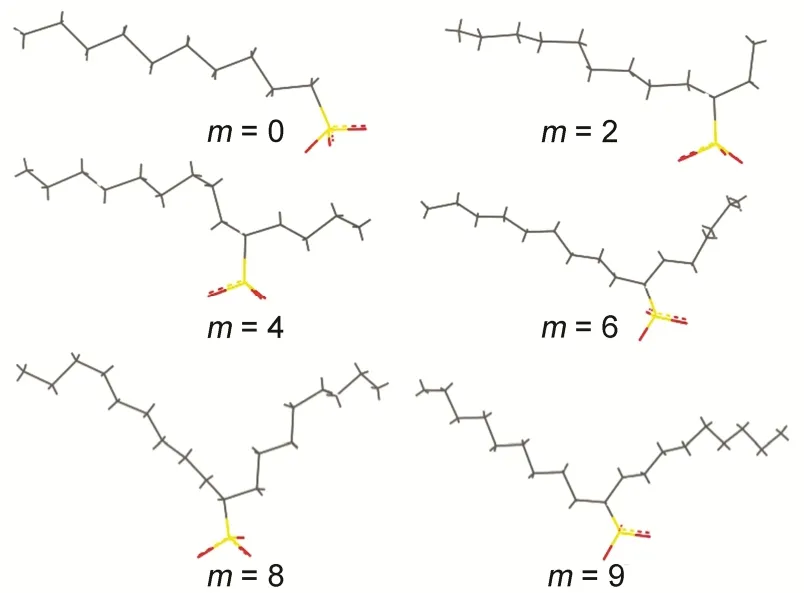
图5 模拟平衡时在界面的结构状态Fig.5 Balanced chemical structures ofThe carbon atoms (gray), sulfur atoms (yellow) and oxygen atoms (red) are highlighted, hydrogen atoms are not shown. color online
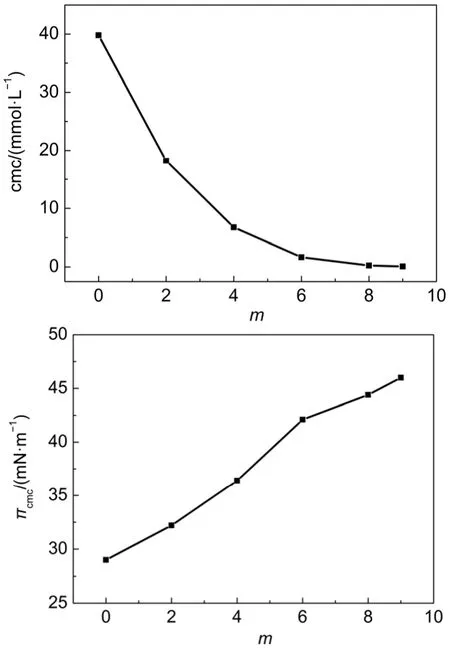
图6 1-Cm-C9-SO3Na的临界胶束浓度及表面压随碳链变化关系 (45oC)34Fig.6 Relation of critical micelle concentration and surface pressure of 1-Cm-C9-SO3Na with the change of carbon chain (45oC)34
根据实验34中得到的1-Cm-C9-SO3Na的临界胶束浓度(cmc)及表面压(π)随烷基碳链变化关系(图6)可知,随着m的增加,其cmc值逐渐降低,降低幅度达到3个数量级(39.8、18.2、6.76、1.66、0.24和0.089 mmol·L-1)。这说明m的增加,表面活性剂由单头单尾变成了单头双尾,临界堆积参数增大,再者碳链增加,其疏水性增强,所以水溶液中更易聚集形成胶束, cmc值降低较为明显。π值是升高的(29.0、32.2、36.4、42.1、44.4和46.0 mN·m-1),这是由于m增加,疏水碳链增多,表面活性剂聚集的致密度增大,所以表面压π值升高。从π值随m值增加的趋势来看,m从0增加到6时,π值增加幅度(3.2、4.2、5.7 mN·m-1)是逐渐增大的,m继续增大,π值的增大幅度(2.3、1.6 mN·m-1)降低。说明m增加到6个和8个的时候,对1-Cm-C9-SO3Na界面聚集行为影响最大,继续增加m,影响较小。1-C0-C9-SO3Na的Krafft点在12oC左右35,36,虽然实验的45oC不同于模拟的25oC,但对于离子型表面活性剂而言,高于Krafft点后,温度对其聚集行为的影响较小,所以用实验的结果来验证我们模拟的结论是有一定的可行性的。
3.2 极性头基间及其与其它组分间的相互作用
上述结果表明,m的增加对表面活性剂的界面聚集行为有明显的影响,会使得表面活性剂远离界面水相区域,也导致Na+的分布随之改变。这些都与体系中组分间的相互作用密切相关。图7给出了各体系中极性头自身间(S-S)、极性头与Na+(S-Na+)和极性头与水分子中的氧原子(S-Ow)的径向分布函数曲线。从图7A和7B都可以看出,m = 4时,曲线的峰值均是最大的,表明此时的极性头基间及其与Na+间的相互作用里最强。所有体系中S-S曲线的出峰位置并非完全一致的,表明m的变化会引起表面活性剂头基间距的改变。图7C表明S-Ow曲线在0.255和0.335 nm处出现两个峰,分别对应于头基的第一和第二水化层。m = 0时,两个峰值均是最大的,表明头基结合水分子数最多,水化能力最强。这也与前面图2A和3A的结果一致,m = 0时,表面活性剂进入水相的部分比其它体系更大。
为了更直观的比较各体系中组分间的相互作用规律,将由各组分间的RDF图得到各个峰的出峰位置以及峰高值列于表3中。从S-S的峰位置可以看出,m = 2,4和6时,其峰位置明显小于其它三者,这表明此时的表面活性剂头基间的距离短,从而导致这三个体系的峰高也基本大于其它体系,尤其是m = 4时,极性头基间的相互作用最强。S-Na+的出峰位置是相同的,数值也与其它体系的模拟结果相一致15。由峰高的变化趋势可以看出,m增加,表面活性剂头基与Na+的相互作用呈现先增大后降低的趋势,m = 4时最大。含有支链比不含支链的要大,这可以从图1中的极性头基的电荷数看出,随着m的增加(从2到9),表面活性剂头基所带负电是逐渐降低,但是都比m = 0时头基所带负电要多。所以对于1-Cm-C9-SO3Na而言,侧链的引入会增大头基的负电性,所以其对Na+的静电吸引力增强,所以含有支链的S-Na+的峰值更高。其中m = 4的峰值最高,表面活性剂头基自身间及其与Na+间的相互作用最大,必然导致头基与水分子的相互作用减弱。
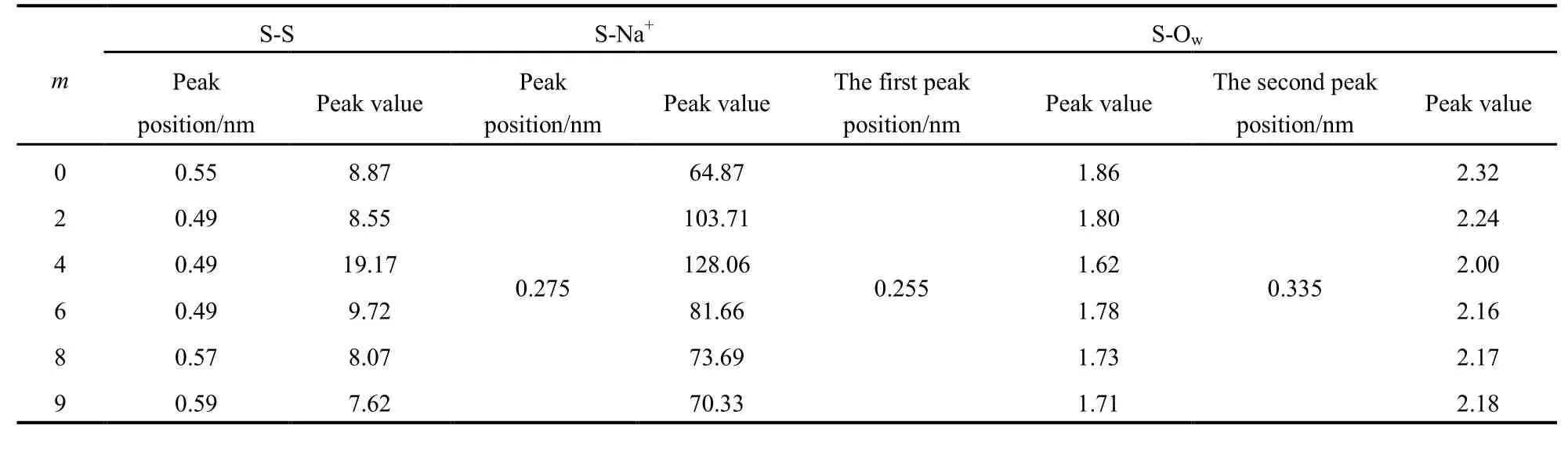
表3 RDFs图中各峰的位置及峰高Table 3 The peak positions and value in the RDFs
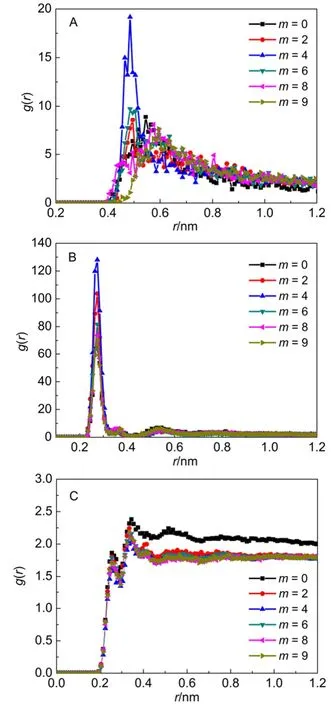
图7 体系中表面活性剂头基间以及与其它组分间的径向分布函数Fig.7 RDF between head group and other components in the system
这从S-Ow的结果就可以明显看出,m = 4时,其第一和第二峰高均是最低的。极性头基的水化能力弱,水化体积小,头基对Na+的静电吸引力就强,所以S-Na+中,其峰值最大。至于1-Cm-C9-SO3Na水化能力的强弱,可以从第一和第二峰位置的峰高看出,m的增加,峰值基本是逐渐降低的,说明表面活性剂头基的水化能力逐渐减弱,表面活性剂的疏水性增强。总而言之,可以发现m = 4时,表面活性剂头基的水化能力最弱,头基与Na+的相互作用最强,此时表面活性剂的聚集体最紧密。
4 结 论
利用分子动力学模拟的方法研究了1-烷基-癸基磺酸钠(1-Cm-C9-SO3Na)在气-液界面的聚集行为,对比了m值的改变对表面活性剂聚集体的影响。模拟发现,随着m的增加,表面活性剂的疏水性增强,其密度分布区域逐渐远离水相。m = 0时,长碳链的倾斜角最大,含有支链之后,受两个碳链间的疏水作用力影响,长碳链更倾向于远离水面,所以其倾斜角减小。
含有支链体系中,m = 2时,支链碳链较短,显刚性,有一定的空间位阻效应,减弱了头基间的相互作用;m = 4时,对表面活性剂聚集行为的影响最大,聚集体紧密度高于其它体系,而且长链和短链的倾斜角均最大,这些现象与实验所得表面压的结果一致,表面压的增加幅度最大。m = 4时,S-Na+和S-S的相互作用最大,但极性头基的水化能力最弱。这主要是由于,支链有一定的柔性,1-C4-C9-SO3Na分子采用平躺的方式在界面上聚集,长、短碳链大致成一条直线,不存在明显的空间位阻效应,且由于此时S-Na+的强相互作用,更多的Na+进入头基附近,降低了头基间的静电排斥,故而头基间距离更小,可以排列的更为紧密;随着m值得进一步增大,支链柔性增加,且与长链的疏水相互作用力大大增强,反而减弱了S-Na+和S-S的相互作用,虽然m继续增大,疏水碳链增加,但是表面活性剂聚集体的致密度并未有明显的增大,表面圧的增加幅度降低。模拟结论表明,当m达到4时,该类双链阴离子表面活性剂的性能达到最优。
(1) Han, D.; Shen, P. P. The Principle and Application of Surfactants in Enhanced Oil Recovery; Petroleum Industry Press: Beijing, 2001, pp 24-27 [韩 东, 沈平平. 表面活性剂驱油原理及应用. 北京:石油工业出版社, 2001, 24-27]
(2) Misra, S. K.; Srivastava, I.; Tripathi, I.; Daza, E.; Ostadhossein, F.; Pan, D. J. Am. Chem. Soc. 2017, 139, 1746. doi: 10.1021/jacs.6b11595
(3) Wedel, B.; Brändel, T.; Bookhold, J.; Hellweg, T. ACS Omega 2017, 2, 84. doi: 10.5414/CN108960
(4) Sandhu, S.; Kumar, R.; Tripathi, N.; Singh, H.; Singh, P.; Kumar, S. Sensor Actuat B-Chem. 2017, 241, 8. doi:10.1016/j.snb.2016.10.041
(5) Guan, Z.; Tang, X. Y.; Nishimura, T.; Huang, Y. M.; Reid, B. J. J. Hazard Mater. 2017, 322, 205. doi: 10.1016/j.jhazmat.2016.02.067
(6) Liu, Z.; Cao, M. W.; Chen, Y.; Fan, Y. X.; Wang, D.; Xu, H.; Wang, Y. L. J. Phys. Chem. B 2016, 120, 4102. doi: 10.1021/acs.jpcb.6b02897
(7) Li, Z. P.; Li, Q. Y. Oilfield Chemistry 1985, 2 (3), 204. [李之平, 李庆莹. 油田化学, 1985, 2 (3), 204.]
(8) Wang, C.; Yan,P.; Xiao, J. X. Acta Phys. -Chim. Sin. 2009, 25 (9), 1775. [王 晨, 严 鹏,肖进新. 物理化学学报, 2009, 25 (9), 1775.] doi: 10.3866/PKU.WHXB20090830
(9) Xu, G. Y.; Luan, Y. X.; Liu, J. Acta Phys. -Chim. Sin. 2005, 21 (4), 450. [徐桂英 栾玉霞,刘 军. 物理化学学报,2005, 21 (4), 450.] doi: 10.3866/PKU.WHXB20050422
(10) De, T. K.; Maitra, A. Adv. Colloid Interface Sci. 1995, 59, 95. doi:10.1016/0001-8686(95)80005-N
(11) Yu, T.; Li, Z.; Ding, W.; Luo, S. Q.; Luan, H. X.; Tong, W.; Qu, G.S.; Cheng, J. C. Acta Phys. -Chim. Sin. 2010, 26 (2), 317. [于 涛, 李钟, 丁 伟, 罗石琼, 栾和鑫, 童 维, 曲广淼, 程杰成. 物理化学学报, 2010, 26 (2), 317.] doi: 10.3866/PKU.WHXB20100224
(12) Yu, T.; Tong, W.; Su Y. B.; Ding, W. Chin. J. Appl. Chem. 2010, 27 (4), 466. [于 涛, 童 维, 宿雅彬, 丁 伟. 应用化学, 2010, 27 (4), 466.] doi: 10.3724/SP.J.1095.2012.00421
(13) Yu, T.; Luo, S. Q.; Ding, W.; Wang, H. M.; Yuan, D. D.; Qu, G. M.; Li, Z. Chin. J. Appl. Chem. 2012, 29 (9), 1060. [于 涛, 罗石琼, 丁伟, 王会敏, 袁丹丹, 曲广淼, 李 钟. 应用化学, 2012, 29 (9), 1060.]
(14) Pang, J. Y.; Zhao, T. T.; Xu, G. Y. J. Surf. Deterg. 2016, 19, 1015. doi: 10.1007/s11743-016-1849-0
(15) Sun, H. Q.; Zhao, T. T.; Cao, X. L.; Yuan, S. L.; Wang, Q. W.; Li, X. S.; Xu, G. Y. Acta Chim. Sin. 2011, 69 (9), 1047. [孙焕泉, 赵涛涛,曹绪龙, 苑世领, 王其伟, 李雪松, 徐桂英. 化学学报, 2011, 69 (9), 1047.]
(16) Griffin, L. R.; Browning, K. L.; Lee, S. Y.; Skoda, M. W. A.; Rogers, S.; Clarke, S. M. Langmuir 2016, 32, 13054. doi: 10.1021/acs.langmuir.6b03601
(17) Yu, Y. C.; Xiao, H. M. Acta Phys. -Chim. Sin. 2009, 25 (1), 30. [于艳春, 肖鹤鸣. 物理化学学报, 2009, 25 (1), 30.] doi: 10.3866/PKU.WHXB20090106
(18) Poghosyan, A. H.; Arsenyan, L. H.; Shahinyan, A. A. J. Surfact. Deterg. 2015, 18, 755. doi:10.1007/s11743-015-1701-y
(19) Poghosyan, A. H.; Arsenyan, L. H.; Shahinyan, A. A. Colloid Polym. Sci. 2014, 292, 3147. doi: 10.1007/s00396-014-3364-z
(20) Hu, S. Q.; Ji, X. J.; Fan, Z. Y.; Zhang, T. T.; Sun, S. Q. Acta Phys. -Chim. Sin. 2015, 31 (1), 83. [胡松青, 纪贤晶, 范忠钰, 张 田, 孙霜青. 物理化学学报, 2015, 31 (1), 83.] doi: 10.3866/PKU.WHXB201411191
(21) You, H.; Zhao, B.; Wang, Z. W. Acta Phys. -Chim. Sin. 2009, 25 (1), 67. [游 慧, 赵 波, 王正武. 物理化学学报, 2009, 25 (1), 67.] doi: 10.3866/PKU.WHXB20090112
(22) Hughes, Z. E.; Walsha, T. R. RSC Advances 2015, 5, 49933. doi: 10.1039/C5RA09192F
(23) Cao, X. L.; He, X. J.; Zhao, G. Q.; Song, X. W.; Wang, Q. W.; Cao, Y. B.; Li, Y. Chem. J. Chin. Univ. 2007, 28 (11), 2106. [曹绪龙, 何秀娟,赵国庆, 宋新旺, 王其伟, 曹嫣镔, 李 英. 高等化学学报, 2007, 28 (11), 2106.]
(24) Ding, W.; Liu, G. Y.; Yu, T.; Qu, G. M.; Cheng, J. C.; Wu, J. Z. Acta Phys. -Chim. Sin. 2010, 26 (3), 727. [丁 伟, 刘国宇, 于 涛, 曲广淼, 程杰成, 吴军政. 物理化学学报, 2010, 26 (3): 727.] doi: 10.3866/PKU.WHXB20100309
(25) Liu, Z. Y.; Wei, N.; Wang, C.; Zhou, H.; Zhang, L.; Liao, Q.; Zhang, L. AIP Advances 2015, 5, 117203. doi: 10.1063/1.4935339
(26) He, X. B.; Guvench, O.; Jr. MacKerell, A. D.; Klein, M. L. J. Phys. Chem. B 2010, 114, 9787. doi: 10.1021/jp101860v
(27) Kabra, V. S.; Gaikar, V. G. J. Mol. Liq. 2008, 142, 143. doi: 10.1016/j.molliq.2008.06.008
(28) Yuan, S. L.; Chen, Y. J.; Xu, G. Y. Colloid Surf. A-Physicochem. Eng. Asp. 2006, 288, 108. doi: 10.1016/j.colsurfa.2006.01.048
(29) Pang, J. Y.; Xu, G. Y. Chem. Phys. Lett. 2012, 537, 118. doi: 10.1016/j.cplett.2012.04.023
(30) Chen, Y. J.; Xu, G. Y.; Yuan, S. L.; Sun, H. Y. Colloids Surf. A 2006, 273,174. doi:10.1016/j.colsurfa.2013.02.026
(31) Pang, J. Y.; Wang, Y. J.; Xu, G. Y.; Han, T. T.; Lv, X.; Zhang, J. J. Phys. Chem. B 2011, 115, 2518. doi: 10.1021/jp110044t
(32) Zhao, T. T.; Xu, G. Y.; Yuan, S. L.; Chen, Y. J.; Yan, H. J. Phys. Chem. B 2010, 114, 5025. doi: 10.1021/jp907438x
(33) Chen, Y. J.; Xu, G. Y. Colloid Surf. A-Physicochem. Eng. Asp. 2013, 424, 26. doi: 10.1016/j.colsurfa.2013.02.026
(34) Granet, R.; Piekarski, S. Colloids Surf. 1988, 33, 321. doi: 10.1016/0166-6622(88)80071-4
(35) Gu, T. R.; Sjöblom, J. Acta Chemica Scandinavica 1991, 45, 762. doi: 10.3891/acta.chem.scand.45-0762
(36) Fekarcha, L.; Tazerouti, A. J. Surf. Deterg. 2012, 15, 419. doi: 10.1007/s11743-012-1335-2
Aggregation Behavior of Double-Chained Anionic Surfactant 1-Cm-C9-SO3Na at Air/Liquid Interface: Molecular Dynamics Simulation
CHEN Yi-Jian1,*ZHOU Hong-Tao1GE Ji-Jiang1XU Gui-Ying2
(1College of Petroleum Engineering, China University of Petroleum (Huadong), Qingdao 266580, Shandong Province, P. R. China;2Key Laboratory of Colloid and Interface Chemistry, Shandong University, Ministry of Education, Jinan 250100, P. R. China)
The aggregation behavior of the double-chained anionic surfactant 1-alkyl-decyl sodium sulfonate (1-Cm-C9-SO3-Na) at the air/liquid interface was investigated using molecular dynamics simulation. The influences of the m value on the interfacial properties of the surfactant were compared using density profile and radial distribution function (RDF). The results showed that the hydrophobic ability of the surfactant increase and the slant angles of hydrophobic carbon chains decrease with increasing m. For m = 4, the 1-C4-C9-SO3Na form aggregates by lying on the interface; the S-S and S-Na+interactions are the highest for m = 4 among all systems studied, while the hydration ability of its polar head is the weakest. The simulation and experimental results show that the interfacial performance is the best for 1-C4-C9-SO3Na.
Molecular dynamics simulation; 1-Alkyl-decyl sodium sulfonate; Air/liquid interface; Aggregation behavior
October 13, 2016; Revised: February 28, 2017; Published online: April 7, 2017.
O641
10.3866/PKU.WHXB201704075
*Corresponding author. Email: chenyijiancs@126.com; Tel: +86-532 86981901.
The project was supported by the Shandong Provincial Natural Science Foundation, China (ZR2014EZ002) and National Natural Science Foundation of China (51574266).
山东省自然科学基金重点项目(ZR2014EZ002)和国家自然科学基金项目(51574266)资助
© Editorial office of Acta Physico-Chimica Sinica
猜你喜欢
分析测试技术与仪器(2022年1期)2022-04-07
数理化解题研究(2020年34期)2021-01-12
河北理科教学研究(2020年2期)2020-09-11
化工管理(2020年5期)2020-01-15
中小学班主任(2019年12期)2019-09-10
天然产物研究与开发(2018年10期)2018-11-06
北京航空航天大学学报(2017年1期)2017-11-24
物理学报(2017年10期)2017-08-09
中国洗涤用品工业(2011年5期)2011-03-20
中学理科·综合版(2008年9期)2008-10-15
