原子转移自由基聚合研究进展
2017-07-06 00:32宋一凡任艳蓉
化学研究 2017年3期
柴 云,宋一凡,任艳蓉,周 慧
(河南大学 化学化工学院,精细化学与工程研究所,河南省阻燃与功能材料工程实验室,河南 开封 475004)
原子转移自由基聚合研究进展
柴 云,宋一凡,任艳蓉,周 慧*
(河南大学 化学化工学院,精细化学与工程研究所,河南省阻燃与功能材料工程实验室,河南 开封 475004)
原子转移自由基聚合(Atom transfer radical polymerization, ATRP)是一种发展较快的可控/活性聚合技术,现已广泛应用于聚合物分子结构设计及众多功能高分子材料的合成. 本文在综述了ATRP 的反应机理的基础上,介绍了引发剂、催化剂、配体、单体等对ATRP的影响,同时综述了降低(或去除)金属盐含量的绿色、高效ATRP聚合体系,如引发剂持续再生活化ATRP,电子转移生成(再生)活化剂ATRP,铁催化体系,光催化体系等. 近年来发展的无金属光诱导的有机催化ATRP聚合体系也做了综述.
原子转移自由基聚合(ATRP);有机催化ATRP;光诱导;活性聚合
传统的自由基聚合反应是一个符合概率统计的随机过程,很难精准控制所得聚合物的组成和结构. 随着高分子研究的不断深入和发展,如高分子应用于自组装及作为光、电、磁功能材料和生物医用材料等,合成具有指定组成和结构的高分子成为高分子合成化学的重要研究领域. SZWARC在无水、无氧等条件下,以萘钠引发苯乙烯聚合,发现不存在链转移和链终止. 于1956年首次提出了“活性聚合物”(Living Polymer)的概念,并确立了活性聚合的技术和方法[1-2]. 其特征在于:
1) 聚合动力学呈现一级动力学行为,即聚合速率与体系中的单体浓度呈线性关系,ln[M]0/[M]对时间t作图应是直线关系,一般来讲链引发速率大于链增长速率;
2) 具有预期的聚合度,即所得聚合物的数均相对分子质量与单体转化率呈线性关系;
3) 所得聚合物的相对分子质量分布符合泊松分布即分布窄,接近于1;
4) 所得聚合物保持活性,即具有再引发单体聚合的能力.
这一聚合技术提供了传统聚合反应所无法提供的手段,使得高分子的分子设计成为现实:1) 通过控制单体和引发剂之间的物质的量之比可以精准合成不同相对分子质量的聚合物;2) 通过顺序加料法可以合成指定结构的多嵌段聚合物;3) 通过合理的结构设计可以得到末端功能化聚合物以及复杂拓扑结构的聚合物(如星形、刷状、超支化、环状聚合物等). 此后人们发展了活性阳离子聚合[3-4],活性开环聚合[5],基团转移聚合[6],极性单体(如甲基丙烯酸甲酯、丙烯酸丁酯)的活性阴离子聚合[7]等. 但上述活性聚合方法存在有聚合反应条件苛刻、聚合工艺流程复杂、难以工业化应用等不足. 同时,上述活性聚合技术的单体覆盖面较窄,主要为苯乙烯、(甲基)丙烯酸酯类等单体,使得分子结构的可设计性较差,除了由阴离子聚合制备的苯乙烯-丁二烯-苯乙烯嵌段共聚物(SBS)和溶液丁苯橡胶实现了工业化以外,其他活性聚合方法很少有工业化应用.
自由基聚合具有单体来源广泛、合成工艺多样、操作简便、容易实现工业化等优点,因此活性/可控自由基聚合的研究与开发更具有实际应用意义. 但自由基聚合的慢引发、快增长、速终止的聚合反应机理决定了聚合产物呈现宽相对分子质量分布,相对分子质量和结构不可控,有时甚至会发生支化、交联等,从而严重影响了聚合物的性能. 因此,如何使自由基聚合具有活性聚合的特征成为当今高分子化学工作者的研究兴趣之一,从活性聚合特征和自由基聚合的机理来分析,实现活性自由基聚合的关键是如何防止聚合过程中因链转移和链终止反应而产生无活性(死)聚合物链. 人们发现通过可逆的链转移或链终止,使活性种(具有链增长活性)和休眠种(暂时无链增长活性)进行快速的可逆转换,可使得聚合体系中自由基浓度控制的很低,便可抑制双基终止,使自由基聚合具有活性聚合的特征. 但这种聚合并不存在真正的无终止,所以不是真正的活性聚合,人们又称这种“活性”自由基聚合为可控自由基聚合. 2010 年国际纯粹与应用化学联合会(IUPAC)推荐将以前的“可控”自由基聚合(“controlled” radical polymerization)或“活性”自由基聚合(“living” radical polymerization)统一称作可逆钝化自由基聚合 (reversible-deactivation radical polymerization (RDRP) or controlled reversible-deactivation radical polymerization)[8].
自从1982年日本学者OTSU等开发了具有引发-转移-终止功能于一身的INIFERTER引发剂,并将其成功地运用到自由基聚合,活性/可控自由基聚合进入一个全新的历史发展时期. 陆续开发出了引发转移终止剂法[9],稳定自由基聚合法或氮氧自由基调控聚合法[10]、原子转移自由基聚合[11-12]、可逆加成-断裂链转移聚合[13]等. INIFERTER方法对聚合过程控制的不是很好,聚合后期动力学行为明显偏离线性关系;相对分子质量分布较宽,限制了在实际中的应用. TEMPO引发体系只适合于苯乙烯及其衍生物的活性聚合,因此工业价值不大. 其中以1995年MATYJASZEWSKI等开发的原子转移自由基聚合(Atom Transfer Radical Polymerization, ATRP)适用单体广泛、反应灵活、反应条件温和等优点成为高分子合成领域最为活跃的前沿课题.
1 发展的第一阶段(1995-2004)
ATRP研究的第一阶段从1995年MATYJASZEWSKI和SAWAMOTO两个课题组几乎同时发表过渡金属催化的活性自由基聚合开始,到致力于开发降低过渡金属含量的绿色聚合方法为止. 这十年的相关研究已有国际顶级学者发表了多篇综述[14-23],现简要总结如下:
1.1 聚合机理的确立
ATRP的聚合机理可以由持续自由基效应(persistent radical effect, PRE)解释,当持续自由基和瞬时自由基在体系中以相同的速率产生的时候,自由基双基终止的产物总是以两种不同自由基交叉形成的交叉产物为优先产物. 过渡金属离子及其卤化合物具有持续性,链自由基作为瞬时自由基,在持续效应作用下,优先与过渡金属化合物作用转移卤素原子,而不是发生双基终止反应[24]. 如图1所示. 在引发阶段,处于低价态的金属配合物 Mtn从有机卤化物 R-X 中夺取卤原子 X,生成自由基 R·和高价态的金属卤化物 Mtn+1-X. 自由基 R·可引发单体聚合,形成链自由基 P·. 而且链自由基P·又可从高价态的金属卤化物 Mtn+1-X 中重新夺取卤原子 X 钝化反应生成形成 R-P-X,并将高价态的金属卤化物还原为低价态配合物 Mtn. 如果 P-X 与 R-X 一样可与 Mtn发生活化反应生成相应的 P·和 Mtn+1-X,同时若P·与 Mtn+1-X 又可反过来发生钝化反应生成 P-X 和 Mtn,则在自由基聚合反应进行的同时,始终伴随着一个自由基活性种与有机大分子卤化物休眠种之间的可逆转移平衡反应. 从本质上看,原子转移自由基聚合实际是一个可逆的催化过程,催化剂Mtn及 Mtn+1-X 的可逆转换控制着聚合体系自由基浓度,使之维持在一个很低的水平.
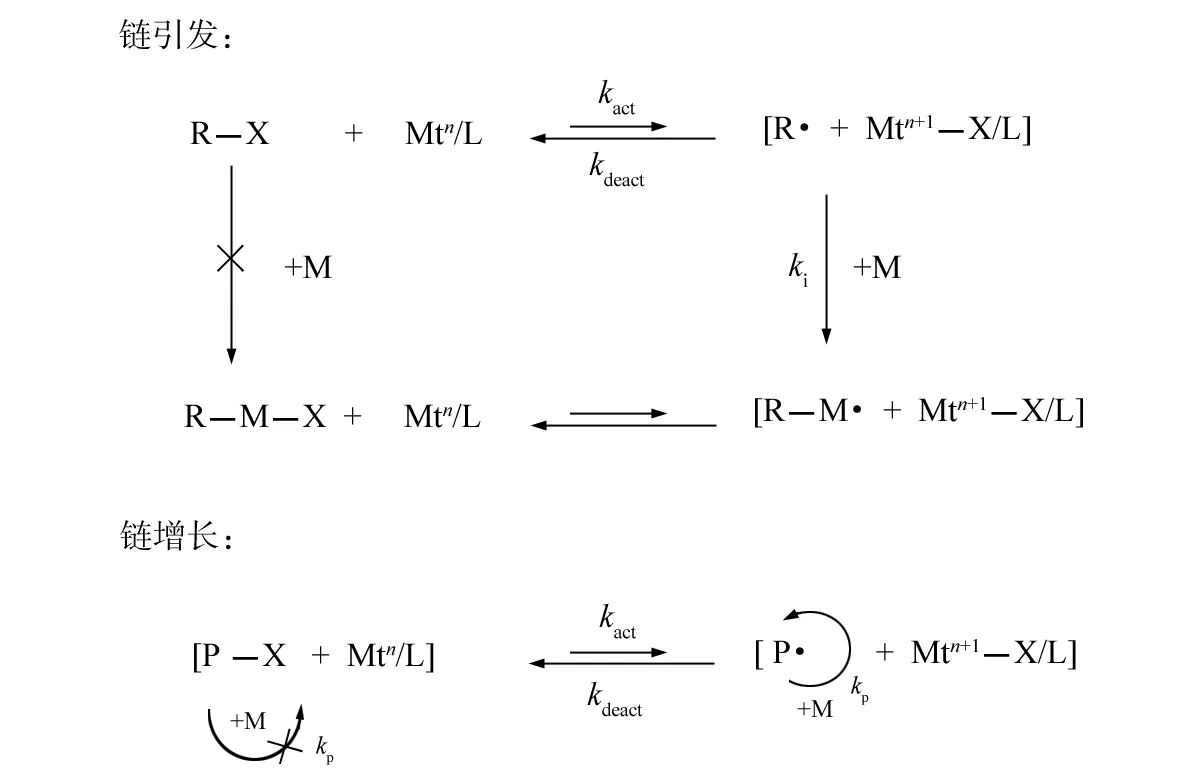
Mtn为低价态过渡金属; Mtn+1为高价态过渡金属; L为有机配体; X为卤素原子.图1 ATRP聚合机理示意图Fig.1 Representative mechanism of ATRP
ATRP 的控制很大程度上依赖于活化过程(产生自由基,kact)与失活过程(形成卤代烃,kdeact)之间恰当的平衡. 其活化速率和失活化速率及平衡常数(KATRP=kact/kdeact)决定了体系中自由基的浓度因此也影响了聚合速率和终止速率,最终影响了聚合物的相对分子质量分布(式1和式2)[25-26].KATRP,kact和kdeact受催化剂、引发剂、单体结构、溶剂的种类以及反应条件等因素的影响. 从机理上探讨这些因素是怎样对三个参数的影响会最终发现更高效的 ATRP 催化体系. 在一个典型的 ATRP 反应中,足够小的KATRP常数会保持体系中的自由基浓度维持在很低的水平,从而降低发生终反应的概率. 另一方面,尽管kdeact远远大于kact,kact和kdeact两个常数都应该足够的大,以用来在一定聚合速度下保持足够小的聚合物相对分子质量分布.

1.2 ATRP体系组成
1.2.1 引发剂
ATRP 引发剂(R-X)在低价金属络合物的活化下均裂产生自由基(R·)并引发单体聚合,同时引发剂中离去基团 X 与低价金属络合物结合形成高价金属络合物失活剂. 引发单体后形成的增长自由基会夺取失活剂上的离去基团 X 形成休眠种,休眠种会在活化剂作用下再次形成增长自由基. 该反复进行的可逆活化/失活过程构成 ATRP 平衡. 因此引发剂必须慎重选择以保证引发过程是定量和快速的,休眠种在聚合体系中是稳定的. 在 ATRP 引发体系中,引发剂的用量和类型,决定着最终产物的相对分子质量及其分布. 因此,选用引发速率快的引发剂可以获得结构规整、相对分子质量分布窄的聚合物. 一般来说,所有α位上含有诱导共轭基团的卤代化合物都能引发 ATRP 反应. 已报道的引发剂有烷基卤化物和苄基卤化物,α-溴代酯,α-卤代酮,α-卤代腈,α-卤酰胺,磺酰卤类化合物等. 通过系统的研究发现:1) 在相同的离去基团 X 下,平衡常数KATRP随着引发剂结构从小到大的顺序为卤代伯碳烷烃 < 卤代仲碳烷烃 < 卤代叔碳烷烃; 2) 对于相同的碳链结构 R 而言,不同卤代化合物的键能是R-Cl > R-Br > R-I,活化速率随着离去基团不同的顺序为I > Br > Cl,因此氯代化合物引发剂率最低,碘代化合物的引发效率最高. 但碘代化合物对光敏感且易与金属形成铬合物,所以,最常见的是溴代化合物作为ATRP的常用引发剂; 3) 在相同级数的碳链结构,相同离去基团情况下,不同取代基结构对引发剂的活性有明显的影响,引发剂活性按取代基结构从大到小的顺序为苯基乙酯基>氰基>苄基>酯基. 根据实验数据和上述的基本结论,MATYJASZEWSKI 课题组对不同的引发剂的平衡常数KATRP进行了排序,α-溴苯乙酸乙酯(Ethylα-bromophenylacetate, EBPA) 是活性最高的引发剂,其活性比苯乙基溴(Phenylethyl bromide, PEBr)高10 000倍,比α-溴丙酸甲酯(Methylα-bromopropionate, MBrP)高100 000倍,如图2所示.

图2 常见引发剂的平衡常数Fig.2 KATRP of common initiator
在烷基卤化物中,四氯化碳是最早被用来作为 ATRP 引发剂[11]. SAWAMOTO 采用CCl4/RuCl2(PPh3)3/MeAl(ODBP)2组成的引发体系首次报道了甲基丙烯酸甲酯(MMA)的 ATRP. 聚合物的相对分子质量随单体的转化率的提高而线性增长,且由 MMA 和 CCl4的投料比来决定,符合一个 CCl4分子产生一个活性聚合物链的假设. 但存在引发效率低,自由基转移等问题.
苄基卤化物由于其产生的苄基自由基具有和苯乙烯类单体增长链自由基相类似的结构,故而被广泛用作苯乙烯类单体的引发剂. 氯化苄和溴化苄(产生的一级碳自由基)由于具有较强的 C-X(X = Cl, Br)键,其引发速率相对较慢. 而 1-苯基卤乙烷由于形成二级碳自由基(和苯乙烯类单体增长链自由基一致),特别适合作为苯乙烯类单体的 ATRP 引发剂. 例如,MATYJASZEWSKI 等采用 1-苯基氯乙烷/CuCl/2,2′-联吡啶(2,2′-bipyridine, bpy)组成的引发体系首次报道了苯乙烯的 ATRP[12]. 所得到的聚苯乙烯相对分子质量在 10 万以内的范围内和理论相对分子质量符合很好,但 PDI 较宽(~1.4). 但当采用1-苯基溴乙烷/CuBr/4,4′-二(1-丁基戊基)-2,2′-联吡啶(4,4′-di(5-nonyl)-2,2′-dipyridyl, dNbpy)取代上述引发体系后,聚苯乙烯的PDI可降至1.1左右.α-卤代酮一般用于Ru催化或者Ni催化的 ATRP. 由于α-卤代酮形成的自由基在反应过程中可能会被 Cu 还原成负离子,导致聚合失控而不太适合作为 Cu 催化的 ATRP 引发剂. 用 N,N-二烷基取代的α-溴酰胺对丙烯酰胺类单体相对来说是一个比较好的引发剂. 例如,SAWAMOTO 等采用 N,N-二甲基-α-溴丙酰胺为引发剂,在 RuCl2(PPh3)3/Al(OiPr)3存在下催化 N,N-二甲基-丙烯酰胺聚合,所得到得聚合物相对分子质量分布较宽(PDI~1.6)但相对分子质量可控;而采用N,N-二甲基-α-氯丙酰胺为引发剂时则聚合物的相对分子质量不可控[27]. MANDAL 等采用没有被烷基取代的α-溴丙酰胺和α-氯丙酰胺为引发剂,在 CuX(X = Br, Cl)/bpy 存在下催化丙烯酰胺聚合,所得到得聚合物相对分子质量分布较宽(PDI>1.6),但聚合物的相对分子质量随转化率的提高而增长,也能进行扩连反应,说明该引发体系对丙烯酰胺还是具有一定的控制性[28].
相对以上几类引发剂而言,α-溴代酯由于其结构中的酯基的吸电子能力中等,使得所产生的自由基的亲电子性能不是太强,故其适合的单体种类广泛,包括苯乙烯类、丙烯酸酯类、甲基丙烯酸酯类等. 其中,由于 2-溴异丁酸乙酯(Ethyl 2-bromoisobutyrate, EBriB)简单易得,引发效率高,适用单体面广,目前已经成为应用最为广泛的 ATRP 引发剂. 例如以 EBriB为引发剂,MMA 为单体,分别采用 Ru,Fe,Cu,Ni等金属盐为催化剂都能得到可控性好的 PMMA. 同时 EBriB 也适合用作 Ru,Fe,Cu,Ni等催化苯乙烯和丙烯酸酯类的引发剂. 另外,自从 Percec 等首次将不同对位取代基 Y (Y = OCH3, H, Cl, NO2, F, CH3)的苯磺酰氯引发苯乙烯的 ATRP 以来[29],发现苯磺酰氯类引发剂和 EBriB 一样,也是一种通用的 ATRP 引发剂,它同样适合苯乙烯类、丙烯酸酯类以及甲基丙烯酸酯类单体的可控聚合[30]. PERCEC 课题组将磺酰氯类引发剂拓展到了芳基磺酰溴、碘类化合物,并成功引发了苯乙烯、丙烯酸甲酯(MA)和甲基丙烯酸甲酯(MMA)的 ATRP 聚合[31-32]. 随着研究的进一步深入,还有一些非常规的 ATRP 引发剂被发现. 例如,ZHANG等[33]发现 N-溴代琥珀酰亚胺(NBS)在 CuBr/bpy 的存在下,可以成功引发甲基丙烯酸甲酯和苯乙烯的ATRP 聚合,得到窄相对分子质量分布的聚合物,但是由于氢消除反应的存在,聚合反应的表观引发效率比较低. PERCEC 课题组将含有 N-Cl 键的化合物产生 N 为活性中心的自由基用于 ATRP 也获得了成功[34].
1.2.2 ATRP 配体
原子转移自由基聚合的催化剂由金属离子和相对应的配体组成. 配体在 ATRP 催化体系中有两个方面的作用:1) 增加过渡金属盐在有机介质中的溶解度; 2) 通过调节催化金属中心原子的氧化还原电势使其具有合适的原子转移活性. 目前报道较多的配体主要有以下三类:1) 含 N 的配体 (吡啶类和胺类),2) 含磷的配体,3) 其他类配体等,常见含氮配体的平衡常数如图3所示.
其中含 N 类配体用得最多,自从第一例 ATRP 报道时采用 2,2′-联吡啶(bpy)用作配体以来,发展到各种不同结构的胺类,包括直链胺类和环状胺类等. MATYJASZEWSKI 等为增进卤化亚铜在聚合体系中的溶解性,在配体 bpy 的 4,4′-位上引入可溶性的侧链(至少含有 4 个碳的烷基链才能满足这一要求). 他们利用 4,4′-二-特丁基-2,2′-联吡啶(dT-bpy)、4,4′-二-正庚基-2,2′-联吡啶(dHbpy)、4,4′-二(5-壬基)-2,2′-联吡啶(dNbpy)代替联吡啶,实现了均相的 ATRP,所得的 PS 和聚(甲基)丙烯酸酯聚合物的 PDI 值明显降低. AMASS[35]通过改变 N-烷基-2-吡啶基亚甲胺配位剂上烷基取代基的长度,使反应体系均相化,提出均相化的体系比非均相化的可控性好,且在极性大的体系比在极性小的体系中要好. ZHANG 等[36]采用 N-己基-2-吡啶基亚甲胺为配位剂成功地进行了 MMA 的均相 ATRP. 含 P 类配体对 Cu 体系效果不好,但可用于其它金属如 Ni、Fe、Rh、Ru和 Re等为催化中心金属原子的配体. 一般来说,配体结构上烷基链越长,其油溶性就越强,越容易使催化体系在油溶性单体里更好地均相化. 还有一些有机酸,如均苯四甲酸,亚氨基二乙酸,丁二酸,异酞酸等适合作为 Fe 盐的配体,能比较好地催化苯乙烯、甲基丙烯酸甲酯和丙烯腈等单体的 ATRP. 另外,还有一些嗡盐也可用作铁盐的高效配体[37-38].
1.2.3 ATRP催化剂
催化剂的作用往往是在配体的作用下形成金属盐配合物,通过金属盐配合物之间的氧化还原反应,决定原子转移自由基聚合中休眠种和活性种之间的可逆动态交换,从而控制 ATRP 反应体系中的自由基浓度. 一个高效的催化剂要满足以下几个必要条件:
1) 金属中心必须至少有两个易达到的稳定氧化态;
2) 金属离子应该对(假)卤素有一定的亲和力;
3) 金属周围的配位空间在氧化反应时能够扩充从而选择性地容纳一个(假)卤素;
4) 配体与金属离子之间的络合作用相对较强.
到目前为止,已经成功开发出许多高效的 ATRP 催化剂, 包括了铜、钌、铁、镍、铑、钴、钼和铼等金属盐.
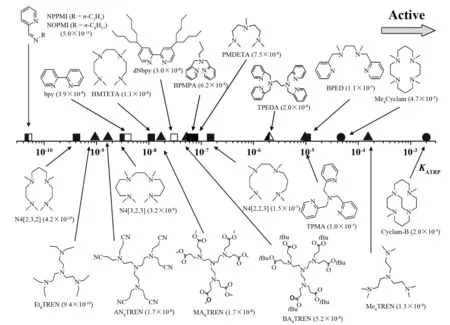
图3 含氮配体的平衡常数KATRPFig.3 KATRP of common ligand contain N
1.2.3.1 铜催化剂
自从MATYJASZEWSKI等在1995年报道首例采用CuCl作为催化剂的ATRP以来,在 ATRP 的所有金属催化剂中,铜盐由于具有很好的催化活性、价廉并且易处理等特点,一直是应用最为广泛的催化剂. 适合于铜盐的配体大多为含 N 配体. 主要使用的配体种类有 2,2′-联吡啶及其衍生物,吡啶亚胺以及其衍生物,三齿、四齿和六齿的线性胺类或环形胺类等. 这些配体的活性次序为联吡啶 < 吡啶亚胺 < 多齿胺 < 三脚架胺 < 环状胺. 邻二氮菲据报道也是铜催化体系的有效配体[39]. BRAR 等[40]报道使用四甲基胍基-三(2-乙基胺)胺(tetramethylguanidino-tris(2-aminoethyl)amine, TMG3-TREN)作为配体与溴化亚铜组成了 ATRP 的催化剂,并用于催化甲基丙烯酸甲酯、丙烯酸正丁酯、苯乙烯和丙烯腈的聚合,并能较好控制上述单体的聚合. DING 等[41]合成了 N,N-二(吡啶基-2-甲基-3-庚氧代基-3-氧代丙基)乙烷-1,2-二胺 (BPED),并把它作为铜体系的配体催化了丙烯酸甲酯、苯乙烯和甲基丙烯酸甲酯的 ATRP 聚合. 结果发现,BPED 与其他线性多齿胺配体相比能够显著提高活化反应的活性. 到目前为止,已经发现了一些高效的含 N 配体. 例如,CuBr/三[2-(二甲胺)乙基]胺 (Tris(2-(dimethylamino)ethyl) amine, Me6TREN) 是一个比较有效的催化剂,它能使丙烯酸酯类单体的聚合反应在室温进行[42]. MATYJASZEWSKI 等合成了一个桥联含 N 配体 Dimethyl cross-bridged cyclam(DMCBCy)[43]和 CuCl 组成的催化体系,其平衡常数是 CuCl/Me6TREN的 30 倍,是至今发现的最有效的配体之一,它甚至可以在 30 ℃快速催化丙烯酸正丁酯的可控聚合. 另外发现的一个六齿含 N 配体(TPEN),它甚至在 CuBr/引发剂=0.005 时可以很好地催化苯乙烯、丙烯酸甲酯和甲基丙烯酸甲酯,是一个极为高效的配体[44].
而在铜盐催化剂中常用的为 CuCl 或者 CuBr,采用 Cu(OAc)为催化剂时,聚合反应速度相比卤化亚铜时较慢,但当在反应体系中加入少量卤化亚铜时则在提高聚合反应速度的同时反应的控制性也加强[45]. CuSCN 和 CuY (Y=O, S, Se)也用作催化剂用于苯乙烯、丙烯酸甲酯和甲基丙烯酸甲酯的 ATRP,也取得了不错的效果[46].
1.2.3.2 钌催化剂
SAWAMOTO 课题组围绕钌催化剂作了大量的研究工作,并就此进行了很好的综述[18]. Ru(Ⅱ)由于具有较大的配位空间能和多种配体配位催化 ATRP. 在 1995 年的第一例钌催化的 ATRP采用 CCl4/RuCl2/PPh3形成的配合物引发 MMA 的聚合时,需有 MeAl (ODBP)2(ODBP = 2,6-二-叔丁基苯酚盐)作为助催化剂,且催化剂的用量较大,所以该引发体系的活性不高. 随后他们把亲水性的苯磺酸钠基团取代 PPh3中的一个苯基可以很好地催化 MMA 以及亲水性单体如甲基丙烯酸羟乙酯(HEMA)的聚合,同时催化剂也很容易除去. TAKAHASHI[47]报道了一种“半茂金属”催化剂Ru(Ind)Cl(PPh3)2(Ind = 茚基), 在 ATRP 反应中对相对分子质量分布控制相对较好. 后来,TAKAHASHI 又报道了另一种钌催化剂 RuH2(PPh3)4,这种催化剂的反应速率较快,如果加入一定的添加剂如n-Bu2NH 则会显著加快反应速率. SIMAL 等则采用含有p-cymene(4-异丙基甲苯)的 RuCl2(p-cymene)的催化剂催化 ATRP,并研究了不同的配体对聚合的影响[48].
1.2.3.3 铁催化剂
铁盐具有价格低、毒性小、生物相容性好等特点,使之在催化合成生物医用高分子材料方面具有特别的吸引力. 所以铁离子与合适的配体络合形成金属络合物催化ATRP 聚合的研究得到了各国学者广泛的关注. 铁催化体系也是目前研究得较多的一种体系. SAWAMOTO 课题组于 1997 年首次报道以卤代羰基化合物为引发剂,FeCl2/PPh3催化的甲基丙烯酸甲酯的可控自由基聚合[49]. 随后,其他学者们又相继开发出了更多高效铁催化剂. 用于铁催化体系的配体主要有三苯基膦及其衍生物,三烷基胺,半茂金属羧基,α-二亚胺,嗡盐,有机羧酸类等. GIBSON等[50]报道了一种高效的配体-三齿水杨酰亚胺(SML). 该配体与氯化亚铁配位后可以形成高效的铁催化剂. 用该催化剂进行苯乙烯的原子转移自由基聚合,得到的聚苯乙烯相对分子质量分布窄至 1.07,这是迄今为止所发现的铁催化剂中最为有效的一种. SCHUBERT 等首次将原来用于铜催化体系的吡啶亚胺类配体用于铁催化体系并取得了成功. 经过优化后,用溴(氯)化亚铁/N-烷基-2-吡啶基-甲酰亚胺催化的甲基丙烯酸甲酯得到的聚甲基丙烯酸甲酯相对分子质量分布保持在 1.35 左右,但实验相对分子质量高于理论值,说明引发效率较低[51]. IBRAHIM 等报道用含喹啉基的四齿配体与氯化亚铁络合形成的铁催化剂可以催化甲基丙烯酸甲酯的原子转移自由基溶液聚合,聚合物相对分子质量分布在 1.27~1.89 之间[52].
1.2.3.4 其他催化剂
实际上,除了铜、钌、铁催化剂外,其他许多过渡金属络合物都可以用作 ATRP的催化剂,如镍、钼、锰、钴、铑和钯催化剂. 镍催化剂能与膦配位,如 Ni(PPh3)Br2或 Ni(PBu3)Br2可用于甲基丙烯酸甲酯和甲基丙烯酸正丁酯的 ATRP 反应. 前者由于热稳定性和溶解性好,可以在低催化剂浓度下控制聚合反应得到高相对分子质量的聚合物. 铑络合物易溶于水,但价格昂贵,所以在ATRP 反应中没有广泛使用. 钯催化剂只能用于甲基丙烯酸甲酯的 ATRP 反应,得到的聚合物相对分子质量分布基本在 1.8 左右,但是它不能用于苯乙烯和丙烯酸酯的聚合,应用单体面太窄. 除了以上的催化体系,原位生成的钼酸(V)锂也可用于苯乙烯的 ATRP反应,可能由于络合物对空气太敏感,所以聚合反应的可控性较差. 选用三价钼盐CpMo(PMe3)2Cl2为催化剂可以得到相对较好的聚合效果. 二茂钴可以较好控制甲基丙烯酸甲酯的 ATRP 聚合,聚合物相对分子质量分布窄,不过聚合反应的引发效率不高,可能是由于二茂钴在催化聚合反应的同时也与自由基发生了副反应. KOUMURA 等发现双核羰基锰 Mn(CO)10是一个光敏感性的催化剂,它不但可以用于丙烯酸甲酯和苯乙烯的聚合,而且还可以得到与醋酸乙烯酯的共聚物[53].
1.2.4 单体
ATRP 中单体结构对聚合控制性的影响比较大,这是因为 ATRP过程是通过可逆活化/失活卤原子与单体单元之间的碳卤键完成的,单体结构很大程度上影响碳卤键的键能. 休眠种中碳卤键的键能取决于单体单元的共轭程度,共轭性越强的单体单元产生的自由基越稳定,碳卤键键能越小,碳卤键越容易均裂,聚合控制性越好. 绝大部分乙烯类单体均适用于 ATRP 过程,如甲基丙烯酸酯类、丙烯酸酯类、苯乙烯类、丙烯腈类和丙烯酰胺类共轭单体,这类单体的双键上取代基中含有极性基团,该极性基团可以稳定生成的单体自由基;无极性基团的二烯类单体(1, 3-丁二烯)则不适用 ATRP. 非共轭乙烯类单体(醋酸乙烯酯等)的 ATRP 过程却比较难实现,这主要是因为金属催化剂很难有效的活化体系中的休眠种成增长自由基.
1.3 其他类型ATRP
1.3.1 反向ATRP
MATYJASZEWSKI 等在 1995 年首次报道铜盐催化的 ATRP 之后不久,以常用的自由基引发剂偶氮二异丁腈(2,2′-azobis(isobutyronitrile), AIBN)和CuCl2/bpy 构成三元引发体系,首次报道了反向 ATRP(Reverse ATRP)[54],得到了窄分布的聚苯乙烯(PDI≈1.3). 但该反应体系是一个非均相反应体系,需要 CuCl2的用量是 AIBN 的 10 倍才能较好地控制聚合,而且反应进行得很缓慢. 这种非均相的反向 ATRP 对 MA 和 MMA 的聚合难以控制. 使用微量的 AIBN,并加入适量的卤化物(2-氯丙腈)作为共引发剂可以解决这一问题. 但由于反向 ATRP 所使用的起始催化剂为氧化态的过渡金属盐,容易保存和操作,随后 MATYJASZEWSKI 等利用烷基取代的 4,4′-二(5-壬基)-2,2′-联吡啶代替 2,2′-联吡啶,实现了 St、MA 和 MMA的均相的反向 ATRP,很好地控制了聚合反应,得到了窄分布的聚合物,解决了反应体系均相化的问题,同时使催化剂用量也相应地降了下来. MOINEAU等[55]则研究了铁盐催化下的反向 ATRP,在 AIBN/FeCl3/PPh3引发下,研究了 MMA 本体及溶液聚合,得到了相对分子质量较高、PDI 窄的聚合物. ZHU 等[56]则采用间苯二甲酸为配位体,报道了 Fe(III)催化的 MMA 的反向 ATRP. DAN等在AIBN/CuBr2/N,N,N′,N″,N″-五甲基二乙烯基三胺 (Pentamethyldiethylenetriamine, PMDETA) 引发的苯乙烯的聚合反应体系中,研究了二甲苯/DMF 和二甲苯/乙醇这两种混合溶剂对聚合反应的影响,结果发现极性溶剂 DMF 的加入使反应体系均相化,同时聚合反应的控制性也得到了加强,但反应有点偏离一级动力学;当在二甲苯中加入一定量的乙醇时,也可以使反应体系有较好的溶解性,且聚合反应的诱导期基本消失[57]. 朱秀林等则首次把微波辐射技术引入到把铜盐和铁盐催化的反向 ATRP 体系中,研究了 MMA 的反应动力学,发现微波的引入在保持“活性”自由基聚合特征的同时还大大加快了聚合反应的速度. CHEN 等则把 AIBN/CuBr2/PMDETA 引发体系引入到离子液体中,并研究了丙烯酸乙酯的反向 ATRP,发现离子液体和催化剂都可回收利用且对聚合反应活性没有太大的影响[58].
在反向 ATRP 中,除了采用 AIBN 作为普通自由基引发剂之外,还有一些采用其他自由基引发剂代替 AIBN 的报道. 其中 BPO 是使用较多的另一种反向ATRP 引发剂,但由于 BPO 与一价铜能进行反应,其机理与 AIBN 不同. QIU等[59]报道了 BPO/Cu(S2CNEt2)/bpy 引发的 MMA的非均相反向 ATRP,可得到窄分布的 PMMA.
1.3.2 正向反向共存ATRP
一般地,对一个成功的反向 ATRP 而言,过渡金属盐的摩尔用量要大于自由基引发剂的摩尔用量,故反应体系中需要加入较大的催化剂用量. 另外,为了使加入体系中的自由基引发剂快速分解,一般需要在高温(≥100 ℃)下进行. 这使反向 ATRP 在克服正向 ATRP 的缺点的同时,又使反向 ATRP 的应用受到限制. 正向反向共存ATRP(Simultaneous Reverse and Normal Initiation ATRP, SR & NI ATRP) 就是在结合了正向和反向ATRP的基础上建立起来的一种新方法[60].
在 SR & NI ATRP 体系中,采用氧化态过渡金属络合物与传统自由基引发剂及ATRP 引发剂并用的双引发体系. 首先传统自由基引发剂(如 AIBN)通过热引发产生自由基 I·,自由基 I·与氧化态的过渡金属盐 X-Mtn+1/L 发生氧化还原反应,并生成还原态的过渡金属络合物 Mtn/L. 加入的 ATRP 引发剂可快速地与该还原态的过渡金属络合物 Mtn/L 进行氧化还原反应生成自由基 R·并引发单体聚合形成增长链自由基R-Pn·,并被氧化态的过渡金属络合物可逆休眠成 R-Pn-X. 同时,由传统自由基产生的引发剂 I-X 也可以形成增长链自由基 I-Pn·,并被氧化态的过渡金属络合物可逆休眠成 I-Pn-X. 需要说明的是,一般地在 SR&NI ATRP 体系中采用比较高效的催化体系,所需的催化剂用量比较低,同时往往控制传统自由基的浓度也很低([I]0/[R-X]0≤10). 这样在整个反应体系中,由普通自由基产生的聚合物链的比例很低,主要还是由 R-X产生的正向 ATRP 产物为主,普通自由基的作用主要是把加入到反应体系中的氧化态的过渡金属盐络合物还原成还原态的 ATRP 催化剂. 2001 年 MATYJASZEWSKI 课题组首次采用氧化态的络合物 CuBr2/Me6-TREN 为催化剂,AIBN 和 2-溴丙酸甲酯(Methyl 3-bromopropionate, MBrP)为双引发剂,[AIBN]0/[MBP]0= 0.06,[AIBN]0/[Cu2+]0=0.5~0.83 下,分别在 60 ℃和 90 ℃下,研究了丙烯酸丁酯(BA)的聚合特点,发现聚合符合一级动力学特征且 PBA 的相对分子质量随转化率的提高而线性增长,同时聚合物的 PDI较窄,显示了很好的“活性”/可控聚合特征. PRAKASH 等沿用同样的思路,以 AIBN/CBr4/Fe(DMF)6](ClO4)3/bpy 组成的引发体系([AIBN]0/[CBr4]0/[Fe(DMF)6](ClO4)3]0/[bpy]0= 0.2/1/0.013/0.04),在 80 ℃下引发了甲基丙烯酸十八酯(SMA)的聚合,PSMA 的相对分子质量随单体转化率的提高而线性增长,但所得到的 PSMA 的 PDI 较宽(1.5~1.75)[61]. MATYJASZEWSKI 课题组还把 SR & NI ATRP 方法应用到水相细乳液聚合体系当中,他们采用 AIBN 或者水溶性引发剂VA-044 (2,2′-azobis(2-(2-imidazolin-2-yl)propane) dihydrochloride)为传统自由基引发剂,单官能团的 MBrP 或者三官能团的三(4-(2-溴代异丁酰氧基)羟基苯基)乙烷 (1,1,1-Tris(4-(2-bromoisobutyryloxy)-phenyl)ethane, TBiBPE)为 ATRP 引发剂,CuBr2为催化剂,BPMODA(bis(2-pyridylmethyl)-octadecylamine),EHA6TREN (tris(2-bis(3-(2-ethylhexoxy)-3-oxo-propyl)aminoethyl)amine)或者 tNtpy(4,4′,4″-tris(5-nonyl)-2,2′terpyridine) 为配体,使用 Brij98 为乳化剂,十六烷为助溶剂,在水相中引发了甲基丙烯酸丁酯(BMA),BA 或者苯乙烯的聚合,成功地得到了可控相对分子质量且 PDI 窄(<1.3)的聚合物[62],随后他们还采用这一方法在水相细乳液聚合体系中合成了线性和三臂的星型嵌段共聚物[63].
2 发展的第二阶段(2005-2014)
相比其他活性自由基聚合方法,ATRP的优点是引发剂、催化剂、配体的多样化,可根据不同的需求进行选择. 但存在如下不足:1)反应条件较苛刻,需在无氧密封条件下进行; 2) 催化剂均为低价态过渡金属化合物,易氧化失活,不利于大批量生产、存储、运输;3) ATRP体系的催化剂、配体用量相对较大(质量分数为单体的的万分之一到千分之一),毒性大且成本高,某些高活性配体合成十分困难(如ME6TREN合成需要四步反应,且产率低);4) 聚合物后处理繁锁,催化剂及配体回收困难. ATRP研究的第二个十年主要集中在开发降低金属盐用量、金属盐的脱除及回收利用、使用无毒或低毒金属盐等绿色、高效的催化体系.
2.1 引发剂持续再生活化ATRP
在上述采用传统自由基引发剂“引发”的 SR & NI ATRP 中,尽管结合了正向和反向ATRP 的各自优势,但还存在着催化剂用量较大的问题. 2006 年 MATYJASZEWSKI课题组在 St/AIBN/EBriB/Cu(Ⅱ)/TPMA, St/EBriB/AIBN/Cu(Ⅱ)/Me6TREN, BA/AIBN/EBriB/Cu(Ⅱ)/TPMA 和 MMA/AIBN/EtBrPA/Cu(Ⅱ)/TPMA 组成的反应体系中,发现如果把催化剂用量降低到5×10-5(质量分数,下同)及以下,同时保持传统自由基引发剂相对于催化剂的用量大大过量(如[AIBN]/[Cu]≥10),将体系中高价态过渡金属不断转换为低价态过渡金属,该反应体系能很好地在较低催化剂用量下控制聚合物的相对分子质量和相对分子质量分布. 他们把这一方法称之为引发剂持续再生活化 ATRP(Initiators for continuous activator regeneration ATRP, ICAR ATRP),其聚合机理如图4所示[64].

图4 ICAR ATRP 聚合机理Fig.4 Polymerization mechanism of ICAR ATRP
该聚合方法和 SR & NI ATRP 相似,但由于在 ICAR ATRP 反应体系中,采用的催化剂用量大大低于慢分解的传统自由基引发剂,使之在整个反应过程中产生的 Cu(Ⅱ)金属络合物可以被过量的传统自由基引发剂不断地还原成 Cu(Ⅰ)催化剂,故使反应体系中的催化剂用量可以大大下降至低于 5×10-5. ICAR ATRP 的聚合动力学显示,聚合反应速率取决于自由基的产生速率,一旦传统自由基引发剂消耗完了以后,聚合反应则很快就会终止[16]. 因为在 ICAR ATRP 体系中自由基引发剂较于催化剂用量大大过量,并且自由基是在整个反应过程中缓慢生成的,ICAR ATRP 的聚合速率取决于自由基的生成速率. 虽然 ICAR ATRP 具有诸多优势,但由于常规自由基引发剂的参与,无可避免地会有一部分常规自由基生成的聚合物链,因此无法用于制备“纯净”的嵌段聚合物.
2.2 电子转移生成活化剂ATRP
2005年MATYJASZEWSKI课题组在SR & NI ATRP的基础上,在反应体系中仍采用常规ATRP中所用的有机卤化物作为引发剂,但采用反向ATRP中所使用的高氧化态的过渡金属盐(如CuCl2)为催化剂. 而常规ATRP中用于产生增长自由基的低氧化态的过渡金属盐(如CuCl)则由加入体系中的高氧化态的过渡金属盐和还原剂进行反应而原位产生. 还原剂通常为多糖类有机化合物(如葡萄糖),抗坏血酸以及辛酸亚锡等易得且无毒或低毒化合物. 同时由于在原位产生低氧化态的过渡金属盐(如CuCl)的反应中,还原剂只和高氧化态的过渡金属盐(如CuCl2)反应而不与体系中的有机卤化物和单体进行反应,这样在原位产生低氧化态的过渡金属盐的过程中就不会影响到有机卤化物和高氧化态的过渡金属盐(如CuCl)之间的反应. 尤其重要的是由于还原剂的存在还可以消耗反应体系中存在的氧气,所以在进行聚合之前加入适量的还原剂(去除消耗氧气的量),整个聚合体系则不必像常规和反向ATRP那样事先要进行除氧,该方法被称之为电子转移生成活化剂ATRP(Activators generated by electron transfer for ATRP, AGET ATRP)[65]. 由于该还原剂只和体系中的氧化态的金属盐Cu(Ⅱ)反应而原位生成催化剂Cu(Ⅰ),但却不像上述的ATRP体系中的AIBN那样产生自由基,因此该反应体系在保留了SR & NI ATRP优点的基础上还克服了SR & NI ATRP在反应体系中不可避免地生成均聚物的弱点,即可得到纯净的嵌段共聚物,一般地,适用于正向 ATRP 的单体,理论上都可适用于 AGET ATRP. 已报道的除了常规典型单体如甲基丙烯酸甲酯、丙烯酸丁酯和苯乙烯之外,还有一些涉及到水溶性如甲基丙烯酸羟乙酯(HEMA)、甲基丙烯酸-N,N-二甲基乙酯(DMAEMA)以及丙烯酰胺等单体AGET ATRP主要由单体、引发剂、包含催化剂和配体的催化体系和还原剂组成. 为了获得一个成功的AGET ATRP,有时候溶剂和温度等因素也应该被充分的考虑. 相比其他ATRP方法,AGET ATRP最大的区别在于引入了不能产生自由基的还原剂来还原ATRP体系中的高价金属. 到目前为止,AGET ATRP常用的还原剂包括辛酸亚锡(Sn(EH)2)、抗坏血酸(As Ac)、葡萄糖、胺、肼、苯酚以及零价金属盐. 采用零价金属丝作为固相还原剂可以方便操作、重复利用并不需要对聚合物进行纯化. 另外,在一些特定的环境中不需要一些额外加入引发剂,一些ATRP单体(如甲基丙烯酸二甲氨基乙酯(DMAEMA))或配体(如TMEDA, HMTETA, PMDETA)可以起到还原剂的作用. 还原剂(如辛酸亚锡、抗坏血酸)也是开环聚合(Ring opening polymerization, ROP)的催化剂,这样可以结合AGET ATRP与ROP通过“一锅法”合成共聚物. 这种方法提供了一种简单的方法在同一个骨架上制备两种不同的聚合物链. MATYJASZEWSKI等利用AGET ATRP和ROP的相结合的方法成功的制备了PCL-b-PODMA,PCL-PBA-PODMA,PLLA-PEOMA和PLA-b-PHEMA等聚合物.
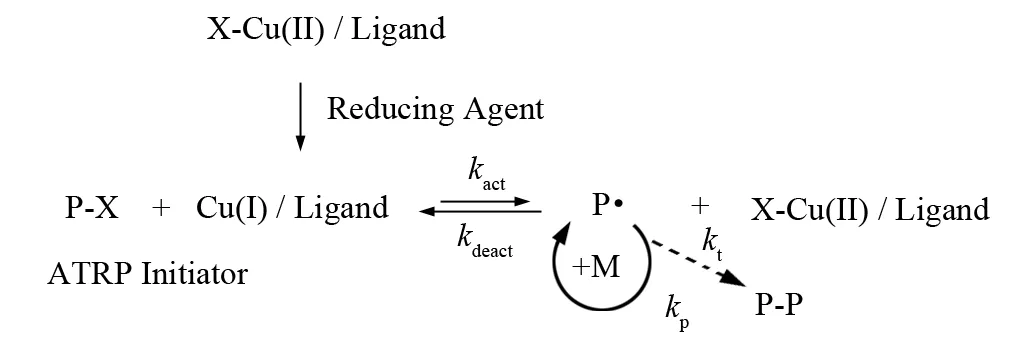
图5 AGET ATRP 聚合机理Fig.5 Polymerization mechanism of AGET ATRP
2.3 电子转移再生活化剂ATRP
MATYJASZEWSKI 课题组于 2006 年在 AGET ATRP 的基础上提出的另一种新的 ATRP 方法——电子转移再生活化剂ATRP(Activators regenerated by electron transfer for ATRP, ARGET ATRP)[66]. 该方法和 AGET ATRP很相似,主要区别在于在过量的还原剂作用下,可以使反应体系中的起始催化剂浓度下降至很低(一般<5×10-5),反应条件相对较温和,可在有少量氧气及自由基存在下进行. 从1.1中公式(1)的聚合速率方程可以看出,ATRP 聚合反应速率取决于链增长速率常数,单体浓度,KATRP,休眠种浓度以及催化剂浓度[X-Cu(Ⅰ)]和失活剂浓度浓度[X-Cu(Ⅱ)]之比. 因此只要维持[X-Cu(Ⅰ)]和 [X-Cu(Ⅱ)]之比不变,即使把加入体系之中的铜盐催化剂的绝对量下降至10-6甚至 10-9数量级也不会影响聚合反应速率. 由该公式可知,要保持ATRP 对聚合物相对分子质量及其分布的可控性,一定量的失活剂浓度总是需要的. 以苯乙烯在 100 ℃时的聚合为例,若要得到 PDI 为 1.2,DPn = 200,转化率p= 90%的聚苯乙烯,需要的失活剂[X-Cu(Ⅱ)]浓度约为 2×10-6. 因此,按此推算在正向 ATRP 中,加入的催化剂的浓度也可以降至很低. 但事实上在正向 ATRP 中,往往需要质量分数在10-4~10-3数量级的催化剂才能比较好地控制聚合反应的进行,这主要是在 ATRP 中不可避免地存在自由基的终止反应,使反应体系中的催化剂[X-Cu(Ⅰ)] 浓度下降. 由上述讨论可知,消耗的催化剂 Cu(Ⅰ)最终转变成失活剂 X-Cu(Ⅱ),若能加入过量的还原剂使失活的催化剂 X-Cu(Ⅱ)能不断地再生成 Cu(Ⅰ),即使在[X-Cu(Ⅰ)]0/[引发剂]0<0.1 的情况下,聚合反应也可以得以进行,所用还原剂还原过程中并不产生新的引发自由基或活性种,所使用催化剂为稳定的高价态过渡金属化合物,有利于大批量生产、存储、运输,该ATRP体系所用的催化剂、配体用量相对较小(Cu类催化剂用量可降至10-6数量级,从而使聚合后处理简单化,所用催化剂还原试剂毒性低(FDA认证)、易得.
2.4 金属盐的脱除
与铜盐配位的配体大多是含氮的极性配体,所得到的催化剂极易溶于甲醇等极性溶剂中,而 ATRP 得到的油溶性聚合物,如 PMMA,PS 等可以在甲醇中析出,根据两者的溶解度差异,通过多次溶解、沉淀循环操作可以把残留的铜催化剂浓度降低到极低的程度[67];以强配体(如乙二胺四乙酸,EDTA)的水溶液洗涤含CuBr2/ PMDETA的 PS 或 PMMA 溶液,可以几乎完全除去聚合物中催化剂,此方法的关键是这种强配体与铜盐的配位能力远大于聚合中所加配体. 由于二氧化硅或氧化铝微粒对金属络合物有很强吸附能力,把含有金属催化剂的聚合物溶液通过中性氧化铝柱可以有效移除聚合物中的金属催化剂,该方法是实验室最常用的方法之一,实验发现催化剂移除效率随着聚合物的极性加大而变低.
MATYJASZEWSKI 等研究发现离子交换柱作为吸附剂也可以很好的移除聚合物中的金属催化剂[68]. 含铜催化剂的聚合物溶液流经含大量-SO3H 的树脂后,铜催化剂可以被有效移除,其移除速率受溶剂极性、温度、树脂种类和催化剂尺寸等因素决定,较高温度和强极性溶液有助于铜催化剂的移除.
利用两相体系高温均相、低温分相的特点,这种特点可以使产物和催化剂之间均相高效反应,异相高效分离. 可用于 ATRP 中金属催化剂分离的液/液两相体系按照两相组成可分为氟/有机两相[69](但含氟试剂和全氟溶剂价格昂贵),离子液体/有机两相,极性/非极性有机两相和水/有机两相[71]. 极性溶剂和非极性溶剂由于极性差异在室温下一般分为液/液两相,而且一些特定组合的溶剂同样具有高温均相,低温分相的特征. 这类溶剂组合有正庚烷/90%乙醇两相,正庚烷/N, N-二甲基乙酰胺两相,聚乙二醇 200/对二甲苯两相等.
通过非共价键负载(物理吸附)[72]、共价键[73]等把催化剂负载到固体表面是一种更有效的分离方法,而且回收的催化剂可以循环使用,节约聚合成本.
2.5 铁催化剂
在可用于ATRP的金属元素中,铁(Fe)因其地壳含量丰富、毒性低而作为一种可实际应用的催化剂. 然而,铁基催化剂与极性溶剂相容性差,限制了很多单体和溶剂的应用.
SAWAMOTO课题组首先报道了一些铁基催化剂(如FeCl2(PPh3)2),或与铁相关的催化体系用于活性自由基聚合. 其他组也报道了与FeX2或者FeX3(X:卤素)相连的多种配体或衍生物,如卤素阴离子(四丁基溴化铵)、极性溶剂(如N-甲基吡咯烷酮)、磷化氢-吡啶杂化配体、高供电子磷化氢配体,等. 最近一个用铁催化剂的研究是基于之前的钌催化二茂铁催化体系的活性自由集聚合衍生出的,用二茂铁癸烷作助催化剂,FeBr2/n-Bu4NBr为引发体系进行反应. 催化效果因二茂铁的氧化还原反应而增强,独立于配体的高催化活性可控制多种极性乙烯基化合物的聚合,如PEGMA,HEMA和 MAA单体. 两种铁复合催化体系的提出尚属首次,未来有望有更多有价值催化体系的发展应用[74-80].
这类复合体系在水中也能表现出高催化活性,将因其固有的生物相容性而在生物材料应用中具有发展前景. MATYJASZEWSKI等通过乙烯基加氢和PEG链段连接,在水中添加还原剂和钠盐促进[(低聚氧化乙烯)甲氧基甲基丙烯酸酯]的活性自由基聚合反应. 在现有复合体系的基础上加以修饰给铁催化活性自由基聚合提供了新思路.
2.6 电化学调控的 ATRP (e ATRP)
在所有的 A(R)GET 和 ICAR ATRP 过程中,还原剂被氧化后会生成副产物:ICAR中会有新的聚合物链,A(R)GET 中会有脱氢抗坏血酸、Sn(IV)物种等. 这些氧化物可能会对聚合物性能有一定的不良影响,也无法对聚合过程实时控制,因此,用电子尤其是电流代替还原剂将很有意义,MATYJASZEWSKI等[81]在电流的作用下将在空气中稳定的CuBr2/Me6TREN成功的还原成CuBr/Me6TREN 来进行 ATRP 控制聚合反应. MATYJASZEWSKI 等将其命名为e ATRP(Electrochemically mediated ATRP)并提出了其聚合机理,如图6所示. 其聚合速率可以通过改变其内在的氧化还原电位(Eapp)实现. 通过增加[CuBr/Me6TREN]/[CuBr2/Me6TREN]的比例使催化体系有一个更负的氧化还原电位,结果使聚合的速率提高. 可喜的是,通过转变氧化还原电位的值,可成功地实现聚合过程的“开”和“关”. 采用10-6级别的金属催化剂,可使聚合物的相对分子质量随转化率的增加而增加,并且最终得到的聚合物端基活性可以维持很高的水平.
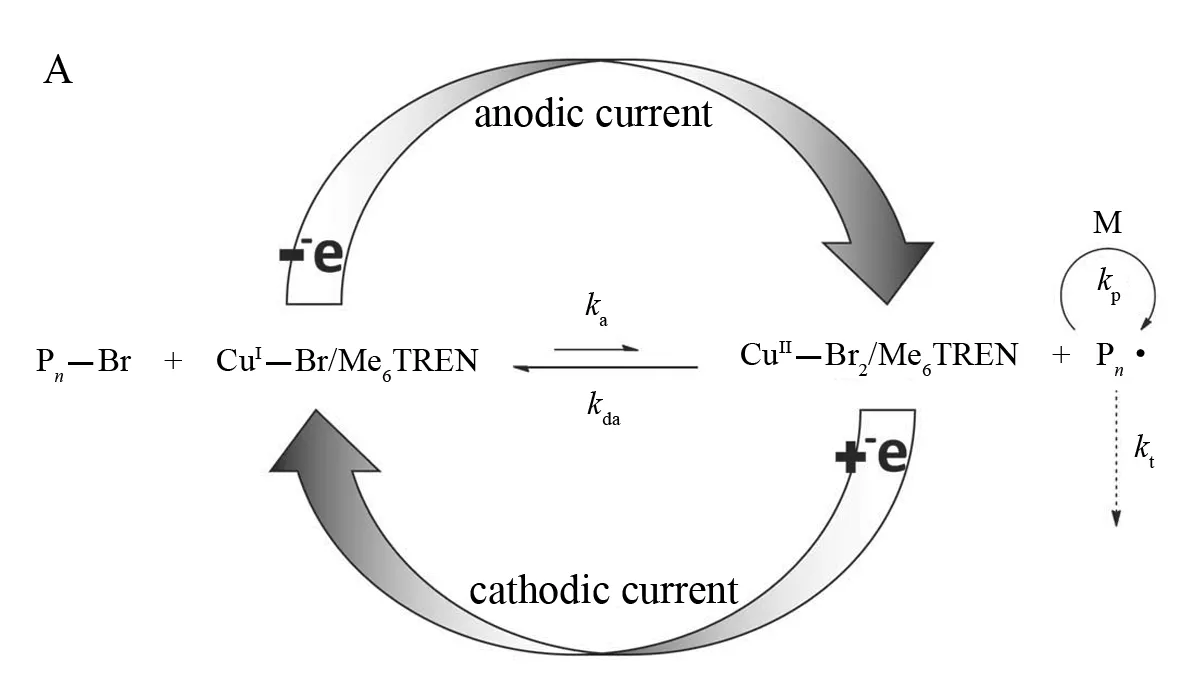
图6 电化学控制的ATRPFig.6 Mechanism of electrochemically mediated ATRP(e ATRP)
2.7 光诱导ATRP聚合
光诱导的“活性”自由基聚合具有温和、高效的特点,聚合可以在室温下进行. 有关光诱导的引发-转移-终止剂聚合(iniferter)[82]、光催化的氮氧稳定自由基聚合(photo-induced NMP)[83-84]、光催化的钴调控自由基聚合[85]、光催化的可逆加成断裂链转移(photo-induced RAFT)聚合[86]已有报道, BOYER 等将光诱导电子转移(PET)概念与RAFT 聚合相结合,提出 PET-RAFT 概念[87],由于 ATRP 在 “活性”自由基聚合中的重要地位,光诱导 ATRP 也被广泛研究.
GUAN 等在 CuCl/bpy 催化 MMA 的 ATRP 中发现有可见光存在时MMA 的聚合速率明显加快,单体转化率几乎可以达到 100%[88]. YAGCI 等发现在光诱导铜催化 ATRP 中催化剂活性种 Cu(Ⅰ)络合物可以在胺类配体存在下直接由高价态 Cu(Ⅱ)络合物还原得到[89]. 在 350 nm 紫外光光照,以溴代丙酸乙酯(EBrP)为引发剂,CuBr2/PMDETA 为催化剂,无外加任何还原剂条件下实现 MMA 的 ATRP,所得PMMA 相对分子质量可控,相对分子质量分布窄;后来,他们发现向该体系中加入甲醇可以进一步提高催化剂的催化活性[90]. MATYJASZEWSKI 课题组结合理论计算和基础实验对活化剂产生方式做了研究[91],研究发现铜盐光催化 ATRP 中,Cu(Ⅰ)活性种主要有以下两种生成方式,(Ⅰ) 在过量胺类配体下 Cu(Ⅱ)络合物直接光化学还原;(Ⅱ) 通过光化学产生自由基去还原生成活化剂,其中第一种形式占主导作用. 关于光催化 ATRP 中如何降低金属催化剂用量的研究方面,MATYJASZEWSKI等发表的以不同波长的发光二极管(LED)作为光源的铜催化 ATRP 体系中铜盐浓度可以有效降低到 10-4[92]. 后来 MOSNACEK 等发现,当以更高活性的引发剂 2-溴丙腈(2-BPN)替代α-溴代异丁酸乙酯(EBriB)时,该体系的铜盐浓度在保证聚合体系控制性的前提下可以进一步降低到 5×10-5[93]. HAWKER 课题组报道了以三(2-苯基吡啶)合铱(fac-[Ir(ppy)3])为光催化剂,EBPA 为高效引发剂的光氧化还原催化MMA 的 ATRP 体系(图7),光催化剂浓度可以降低至5×10-5,聚合具有“开”、“关”性能,并且该体系也适用于丙烯酸酯类单体的聚合[94]. 另外,水相中的10-6铜催化的光诱导 ATRP 体系也被成功开发出来[95],该体系结合了光化学温和、高效和水作溶剂绿色环保的双重优势,然而以毒性较大的铜作催化剂是其不足之处.

图7 Ir(III)配合物催化的光控ATRPFig.7 ATRP mediated by light employing fac-[Ir(ppy)3] as the catalyst
3 发展的第三阶段(2014-)
ATRP 在合成相对分子质量可控、相对分子质量分布窄的拓扑结构聚合物方面展现出强大的适用性,然而 ATRP 中金属催化剂的使用造成最终聚合物的污染一直是限制其大规模应用的一个重要因素,重金属残留不仅加速聚合物的老化,而且在特定领域的应用(如医用材料、电子元件等)还会进一步受限. 前面讨论的 ATRP 体系中都需要金属催化剂的参与,开发无金属催化的 ATRP 体系即有机催化ATRP(Organocatalyzed Atom Transfer Radical Polymerization, O-ATRP)可以从根本上解决金属催化剂带来的不利影响.
2014年美国加州大学圣塔芭芭拉分校的HAWKER课题组联合陶氏化学公司(Dow Chemical Company)的研究人员在以前铱光氧化还原催化剂催化ATRP工作基础上,以 10-苯基吩噻嗪(10-Phenylphenothiazine PTH)为无金属的光氧化还原催化剂,在LED 发出的 380 nm 紫外光辐照下,以 PTH/EBPA 为引发体系的无金属催化 ATRP 中可以实现 MMA,DMAEMA 等单体的“活性”自由基聚合,并通过大分子引发剂的扩链反应证明所得聚合物为活性聚合物[96],其聚合机理如图8所示,该体系可以方便地通过光的“开”和“关”实现聚合过程的“开”和“关”. 该体系所得到的聚合物的 Mn,GPC值与理论值较吻合,且相对分子质量分布在 1.18~1.32 之间,具有明显的“活性”/可控特征. 另外,通过该体系还可以制备控制性较好的嵌段聚合物. 以 PTH 为催化剂的无金属 ATRP 体系也被拓展到丙烯腈的聚合中[97].
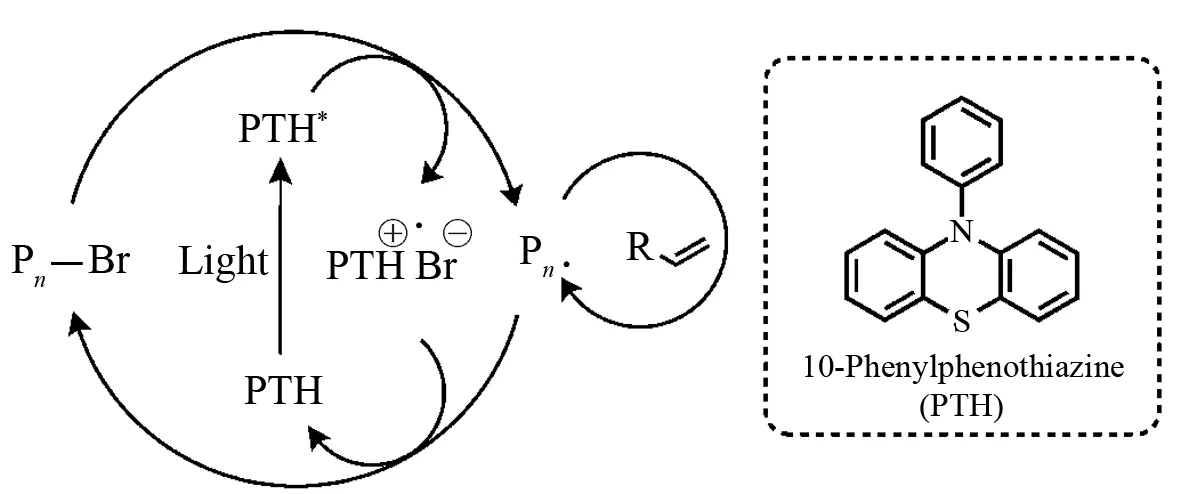
图8 10-苯基吩噻嗪作为催化剂的无金属光催化ATRPFig.8 Metal-free photomediated ATRP with 10-phenylphenothiazine as the catalyst
美国科罗拉多大学波尔德分校的MIYAKE教授小组,几乎同时报道了苝(又名二萘嵌苯,Perylene)有机催化的光控ATRP[98]. 其聚合机理如图9所示.
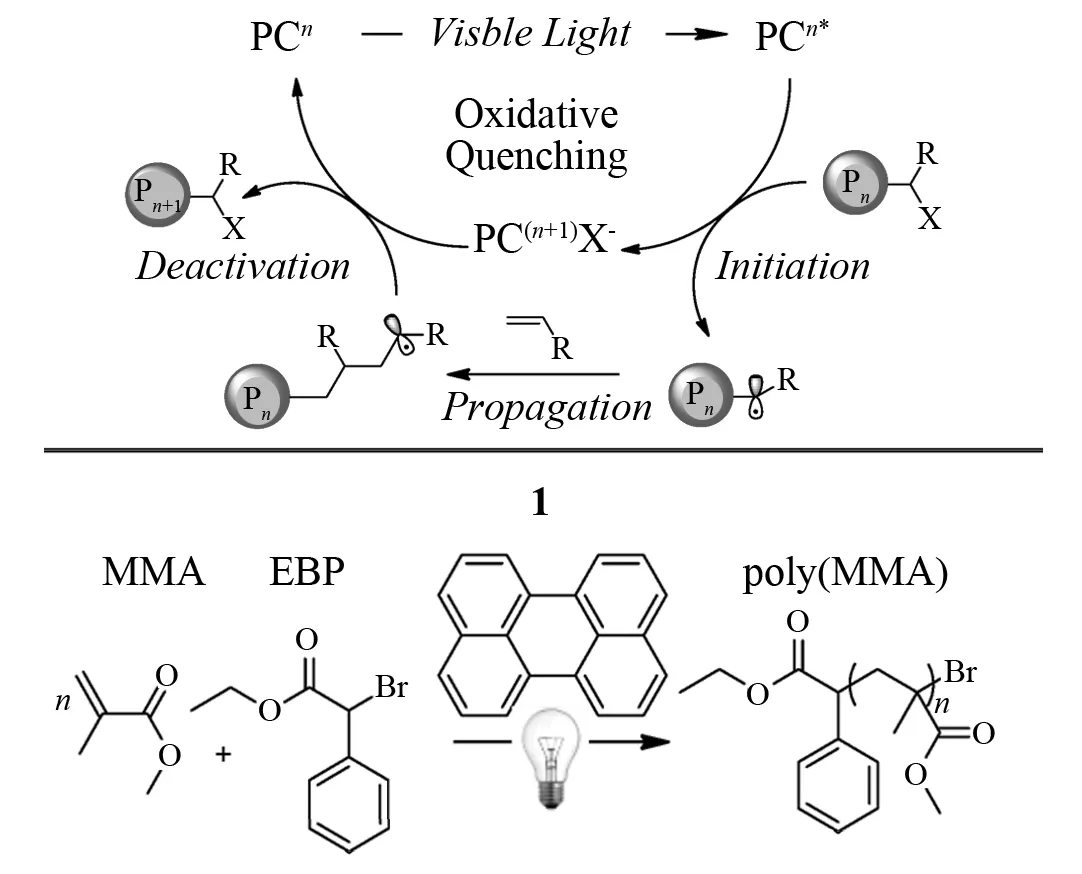
图9 苝作为催化剂的无金属光催化ATRPFig.9 Photoredox-mediated ATRP using perylene as an organic photocatalyst by natural sunligh
两个课题组的报道非常类似:
1)相同的单体(甲基丙烯酸甲酯methyl methacrylate,MMA)和引发剂(α-溴代苯乙酸乙酯);
2) 一级动力学特征;
3) 完美的on-off控制实验;
4) 使用高聚的MMA作为引发剂,可以实现与新单体的嵌段聚合. 不同之处是HAWKER教授的聚合方法相对分子质量偏小,而MIYAKE教授的方法转化率偏低,引发剂效率和PDI很能同时维持较高水平,但苝具有致癌性.
苏州大学朱秀林教授课题组也报道了以荧光素[99]为催化剂在可见光(450 nm)辐照下的无金属 ATRP 过程. 无金属 ATRP 研究才刚刚起步,催化体系催化活性不高,所加的有机染料在聚合物中残留比较严重,而且理论体系还不完善,需要研究者不断地努力.
MIYAKE小组继续在此方向上深入研究,在Science杂志发表文章[100],将催化剂骨架锁定在5,10-二芳基二氢吩嗪,通过密度泛函理论(density functional theory,DFT)计算了4种催化剂以及催化循环各个中间体的氧化还原电势,证实催化循环的有序进行(图10). 实际地筛选发现,催化剂3的效果最好,PDI和引发剂效率可以达1.17和75.9%. 理论计算与实验相结合是该文章的最大特色.

图10 二芳基二氢吩嗪光催化的ATRP聚合机理Fig.10 Prpposed mechanism for ATRP using diphenyl dihydrophenazine as photoredox catalyst
研究小组继续通过DFT的方法分析并总结了激发态催化剂的电子离域规律,不仅解释了催化剂性能差别的内在原因,而且指导合成了更高效的催化剂5和6,PDI和引发剂效率分别为1.23和99.3%,见图12.

图11 光引发剂3的聚合结果Fig.11 Polymerization results using PC 3

图12 光引发剂5和6的理论计算Fig.12 Computationally directed discovery of PCs 5 and 6
4 结论与展望
ATRP的研究已经取得了飞速的发展,开发出了多种聚合体系和方法,但因其重金属残留、引发剂及配体的毒性、成本等因素离工业实际应用有较大的距离. 回顾高分子合成发展史可以知道,Zigler-Natta催化剂催化烯烃聚合的基础研究成果发表不到十年就应用到工业化生产,形成庞大的聚烯烃产业,ATRP的研究则一直停留在实验室研究阶段,若迟迟不能工业化应用,估计这方面的研究会进入平台期. 近几年发展的无金属光催化ATRP聚合体系,即有机催化ATRP为解决重金属残留问题开辟了新的途径. 但仍需要解决光催化剂的成本与残留毒性问题,需要高分子化学工作者继续努力.
[1] SZWARC M. “Living polymers” [J]. Nature, 1956, 178: 1168-1139.
[2] SZWARC M, LEVY M, MILKOVICH R. Polymerization initiated by electron transfer to monomer. A new method of formation of block polymers [J]. Journal of the American Chemical Society, 1956, 78(11): 2656-2657.
[3] MIYAMOTO M, SAWAMOTO M, HIGASHIMURA T. Living polymerization of isobutyl vinyl ether with hydrogen iodide/iodine initiating system [J]. Macromolecules, 1984, 17(3): 265-268.
[4] FAUST R, KENNEDY J P. Living carbocationic polymerization demonstration of the living polymerization of isobutylene [J]. Polymer Bulletin, 1986, 15: 317-323.
[5] AIDA T, INOUE S. Living polymerization of epoxides with metalloporphyrin and synthesis of block copolymers with controlled chain lengths [J]. Macromolecules, 1981, 14(5): 1162-1166.
[6] WEBSTER O W, HERTLER W R, SOGAH D Y. Group-transfer polymerization. 1. A new concept for addition polymerization with organosilicon initiators [J]. Journal of the American Chemical Society, 1983, 105(17): 5706-5708.
[7] FAYT R, FORTE R, JACOBS C, et al. New initiator system for the living anionic polymerization of tert-alkyl acrylates [J]. Macromolecules, 1987, 20(6): 1442-1444.
[8] JENKINS A D, JONES R G, MOAD G. Terminology for reversible-deactivation radical polymerization previously called “controlled” radical or “living” cadical polymerization (IUPAC recommendations 2010) [J]. Pure and Applied Chemistry, 2010, 82(2): 483-491.
[9] OTSU T, YOSHIDA M. Role of initiator-transfer agent-terminator (iniferter) in radical polymerizations: Polymer design by organic disulfides as iniferters [J]. Die Makromolekulare Chemie Rapid Comrnunication, 1982, 3: 127-132.
[10] GEORGE M K, VEREGIN R P N, KAZMAIER P R M, et al. Narrow molecular weight resins by a free-radical polymerization process [J]. Macromolecules, 1993, 26(11): 2987-2988.
[11] KATO M, KAMIGAITO M, SAWAMOTO M, et al. Polymerization of methyl methacrylate with the carbon tetrachloride/dichlorotris-(triphenylphosphine)ruthenium(Ⅱ)/methylaluminum bis(2,6-di-tert-bulphenoxide) initiating system: Possibility of living radical polymerization [J]. Macromolecules, 1995, 28(5): 1721-1723.
[12] WANG J S, MATYJASZEWSKI K. Controlled/“living” radical polymerization. Atom transfer radical polymerization in the presence of transition-mental complexs [J]. Journal of the American Chemical Society, 1995, 117(20): 5614-5615.
[13] CHIEFARI J, CHONG Y K, ERCOLE F, et al. Living free-radical polymerization by reversible addition fragmentation chain transfer: The RAFT process [J]. Macromolecules, 1998, 31(16): 5559-5562.
[14] MATYJASZEWSKI K, XIA J H. Atom transfer radical polymerization [J]. Chemical Reviews, 2001, 101(9): 2921-2990.
[15] KAMIGATO M, ANDO T, SAWAMOTO M, et al. Metal-catalyzed living radical polymerization [J]. Chemical Reviews, 2001, 107(12): 3689-3745.
[16] BRAUNECKER W, MATYJASZEWSKI K. Controlled/living radical polymerization: Features, developments, and perspectives [J]. Progress in Polymer Science, 2007, 32(1): 93-146.
[17] TSAREVSKY N V, MATYJASZEWSKI K. “Green” atom transfer radical polymerization: from process design to preparation of well-defined environmentally friendly polymeric materials [J]. Chemical Reviews, 2007, 107(6): 2270-2299.
[18] TANG W, KWAK Y, BRAUNECKER W, et al. Understanding atom transfer radical polymerization: Effect of ligand and initiator structures on the equilibrium constants [J]. Journal of the American Chemical Society, 2008, 130(32): 10702-10713.
[19] OUCHI M, TERASHIMA T, SAWAMOTO M. Transition metal-catalyzed living radical polymerization: Toward perfection in catalysis and precision polymer synthesis[J]. Chemical Reviews, 2009, 109(11): 4963-5050.
[20] di LENA F, MATYJASZEWSKI K. Transition metal catalysts for controlled radical polymerization [J]. Progress in Polymer Science, 2010, 35(8): 959-1021.
[21] MATYJASZEWSKI K. Atom transfer radical polymerization (ATRP): Current status and future perspectives [J]. Macromolecules, 2012, 45(10): 4015-4039.
[22] SIEGWART D J, OH J K, MATYJASZEWSKI K. ATRP in the design of functional materials for biomedical applications [J]. Progress in Polymer Science, 2012, 37(1): 18- 37.
[23] MATYJASZEWSKI K, TSAREVSKY N V. Macromolecular engineering by atom transfer radical polymerization[J]. Journal of the American Chemical Society, 2014, 136(18): 6513-6533.
[24] Fische H. The persistent radical effect: A principle for selective radical reactions and living radical polymerizations [J]. Chemical Reviews, 2001, 101: 3581-3610.
[25] GOTO A, FUKUDA T. Kinetics of living radical polymerization [J]. Progress in Polymer Science, 2004, 29(4): 329-385.
[26] MASTAN E, LI X H, ZHU S P. Modeling and theoretical development in controlled radical polymerization [J]. Progress in Polymer Science, 2015, 45: 71-101.
[27] SENOO M, KOTANI Y, KAMIGAITO M, et al. Living radical polymerization of N,N-dimethylacrylamide with RuCl2(PPh3)3-based initiating systems [J]. Macromolecules, 1999, 32(24): 8005-8009.
[28] JEWRAJKA S K, MANDAL B M. Living radical polymerization 1. The case of atom transfer radical polymerization of acrylamide in aqueous-based medium [J]. Macromolecules, 2003, 36(22): 311-317.
[29] PERCEC V, BARBOIU B. “Living” radical polymerization of styrene initiated by arenesulfonyl chlorides and Cu/bpy/LCl [J]. Macromolecules, 1995, 28(23): 7970-7972.
[30] PERCEC V, BARBOIU B. “Living” radical polymerization of styrene initiated arenesulfonyl chloride and omides methacrylates, and acrylates [J]. Journal of the American Chemical Society, 1998, 120: 305-316.
[31] GRIGORAS C, PERCEC V. Arenesulfonyl bromides: the second universal class of functional initiators for the metal-catalyzed living radical polymerization of methacrylates, acrylates, and styrenes [J]. Journal of Polymer Science Part A: Polymer Chemitry, 2005, 43: 319-330.
[32] PERCEC V, GRIGORAS C. Arenesulfonyl iodides: The third universal class of functional initiators for the metal-catalyzed living radical polymerization of methacrylates and styrenes [J]. Journal of Polymer Science Part A: Polymer Chemitry, 2005, 43: 3920-3931.
[33] JIANG J G, ZHANG K D, ZHOU H. Atom transfer radical polymerization initiated by N-bromosuccinimide [J]. Journal of Polymer Science Part A: Polymer Chemitry, 2004, 42: 5811-5816.
[34] PERCEC V, GRIGORAS C. N-chloro amides, lactams, carbamates, and imides. new classes of initiators for the metal-catalyzed living radical polymerization of methacrylates [J]. Journal of Polymer Science Part A: Polymer Chemitry, 2005, 43: 5283-5299.
[35] AMASS A J, WYRES C A, COLCLOUGH E, et al. N-alkyl-2-pyridinemethanimine mediated atom transfer radical polymerisation of styrene: the transition from heterogeneous to homogeneous catalysis [J]. Polymer, 2000, 41: 1697-1702.
[36] ZHANG H, KLUMPERMAN B, MING W, et al. Effect of Cu(Ⅱ) on the kinetics of the homogeneous atom transfer radical polymerization of methyl methacrylate [J]. Macromolecules, 2001, 34: 6169-6173.
[37] TEODORESCU M, GAYNOR S G, MATYJASZEWSKI K. Halide anions as ligands in iron-mediated atom transfer radical polymerization [J]. Macromolecules, 2000, 33: 2335-2339.
[38] ISHIO M, KATSUBE M, OUCHI M, et al. Active, versatile, and removable iron catalysts with phosphazenium salts for living radical polymerization of methacrylates [J]. Macromolecules, 2009, 42: 188-193.
[39] DESTARAC M, BESSIERE J M, BOUTEVIN B. transition metal catalyzed atom transfer radical polymerization: from heterogeneous to homogeneous catalysis using 1,10-phenantroline and its deviatives as new copper(Ⅰ) ligands [J]. Macromolecule Rapid Communication, 1997, 18: 967-974.
[40] BRAR A S, KAUR S. Tetramethylguanidino-tris(2-aminoethyl)amine: A Novel ligand for copper-based atom transfer radical polymerization[J]. Journal of Polymer Science Part A: Polymer Chemitry, 2005, 43: 5906-5922.
[41] DING S J, SHEN Y Q, RADOSE M. A new tetradentate ligand or atom transfer radical polymerization [J]. Journal of Polymer Science Part A: Polymer Chemitry, 2004, 42(14): 3553-3562.
[42] QUEFFELEC J, GAYNOR S G, MATYJASZEWSKI K. Optimization of atom transfer radical polymerization using Cu(Ⅰ)/tris(2-(dimethylamino)ethyl)amine as a catalyst [J]. Macromolecules, 2000, 33: 8629-8639.
[43] TSAREVSKY N V, BRAUNECKER W A, MATYJASZEWSKI K. Electron transfer reactions relevant to atom transfer radical polymerization [J]. Journal of Organo-metallic Chemistry, 2007, 692: 3212-3222.
[44] TANG W, KWAK Y, BRAUNECKER W, et al. Understanding atom transfer radical polymerization: Effect of ligand and initiator structures on the equilibrium constants [J]. Journal of the American Chemical Society, 2008, 130: 10702-10713.
[45] MATYJASZEWSKI K, WEI M, XIA J, et al. Atom transfer radical polymerization of styrene catalyzed by copper carboxylate complexes [J]. Macromolecule Chemistry Physics, 1998, 199: 2289-2292.
[46] PERCEC V, ASANDEI A D, ASGARZADEH F, et al. Cu(Ⅰ) and Cu(Ⅰ)I salts of group via elements as catalysts for living radical polymerization initiated with sulfonyl chlorides [J]. Journal of Polymer Science Part A: Polymer Chemitry, 2000, 38(20): 3839-3843.
[47] TAKAHASHI H, ANDO T, KAMIGAITO M, et al. Half-metallocene-type ruthenium complexes as active catalysts for living radical polymerization of methyl methacrylate and styrene [J]. Macromolecules, 1999, 32(11): 3820-3823.
[48] SIMAL F, DEMONCEAU A, NOELS A F. Highly efficient ruthenium-based catalytic systems for the controlled free-radical polymerization of vinyl monomers [J]. Angewandte Chemie International Edition, 1999, 38(4): 538-540.
[49] ANDO T, KAMIGAITO M, SAWAMOTO M. Iron(Ⅱ) chloride complex for living radical polymerization of methyl methacrylate [J]. Macromolecules, 1997, 30: 4507-4510.
[50] O’REILLY R K, GIBSON V C, WHITE A J P, et al. Design of highly active iron-based catalysts for atom transfer radical polymerization: Tridentate salicylaldiminato ligands affording near ideal nernstian behavior [J]. Journal of the American Chemical Society, 2003, 125(28): 8450-8451.
[51] ZHANG H, SCHUBERT U S. An efficient iron-based catalyst bearing N-alkyl-2-pyridylmethanimine ligand for atom transfer radical polymerization [J]. Chemical Communications, 2004, 40(7): 858-859.
[52] IBRAHIM K, YLIHEIKKIL K, ABU-SURRAH A, et al. Polymerization of methyl methacrylate in the presence of Iron(Ⅱ) complex with tetradentate nitrogen ligands under conditions of atom transfer radical polymerization [J]. European Polymer Journal, 2004, 40: 1095-1104.
[53] KOUMURA K, SATOH K, KAMIGAITO M, et al. Manganese-based controlled/living radical polymerization of vinyl acetate, methyl acrylate, and styrene: Highly active, versatile, and photoresponsive systems [J]. Macromolecules, 2008, 41(20): 7359-7367.
[54] WANG J S, MATYJASZEWSKI K. “Living”/controlled radical polymerization. Transition-metal-catalyzed atom transfer radical polymerization in the presence of a conventional radical initiator [J]. Macromolecules, 1995, 28(22): 7572-7573.
[55] MOINEAU G, DUBOIS P, JÉRME R, et al. Alternative atom transfer radical polymerization for mma using FeCl3and AIBN in the presence of triphenylphosphine: An easy way to well-controlled PMMA [J]. Macromolecules, 1998, 31(2): 545-547.
[56] ZHU S, YAN D, ZHANG G. Reverse atom transfer radical polymerization of methyl methacrylate with a new catalytic system, FeCl3/Isophthalic acid [J]. Journal of Polymer Science Part A: Polymer Chemitry, 2001, 39: 765-774.
[57] YI Z, PAN K, JIANG L, et al. Copper-based reverse ATRP process of styrene in mixed solvents [J]. European Polymer Journal, 2007, 43: 2557-2563.
[58] CHEN H, LIANG Y, WANG M, et al. Reverse ATRP of ethyl acrylate with ionic liquids as reaction medium [J]. Chemical Engineering Journal, 2009, 147: 297-301.
[59] LI P, QIU K Y. Copper(Ⅱ) compound catalyzed living radical polymerization of methyl methacrylate in the presence of benzoyl peroxide [J]. Macromolecules, 2002, 35(23): 8906-8908.
[60] GROMADA J, MATYJASZEWSKI K. Simultaneous reverse and normal initiation in atom transfer radical polymerization [J]. Macromolecules, 2001, 34: 7664-7671.
[61] SAIKIA P J, GOSWAMI A, BARUAH S D. Transition metal-catalyzed atom transfer radical polymerization of stearyl methacrylate in the presence of carbon tetrabromide and a conventional radical initiator [J]. Journal Applied Polymer Science, 2002, 86: 386-394.
[62] LI M, MIN K, MATYJASZEWSKI K. ATRP in waterborne miniemulsion via a simultaneous reverse and normal initiation process [J]. Macromolecules, 2004, 37(6): 2106-2112.
[63] LI M, JAHED N M, MIN K, et al. Preparation of linear and star-shaped block copolymers by ATRP using simultaneous reverse and normal initiation process in bulk and miniemulsion [J]. Macromolecules, 2004, 37(7): 2434-2441.
[64] MATYJASZEWSKI K, JAKUBOWSKI W, MIN K, et al. Diminishing catalyst concentration in atom transferradical polymerization with reducing agents [J]. Proceedings of the National Academy of Sciences of the United States of America, 2006,103(42): 15309-15314.
[65] JAKUBOWSKI W, MATYJASZEWSKI K. Activator generated by electron transfer for atom transfer radical polymerization [J]. Macromolecules, 2005, 38(9): 4139-4146.
[66] JAKUBOWSKI W, MIN K, MATYJASZEWSKI K. Activators regenerated by electron transfer for atom transfer radical polymerization of styrene [J]. Macromolecules, 2006, 39(1): 39-45.
[67] KASKO A M, HEINTZ A M, PUGH C. The effect of molecular architecture on the thermotropic behavior of poly[11-(4-cyanophenyl-4-phenoxy)undecyl Acrylate] and its relation to polydispersity [J]. Macromolecules, 1998, 31(2): 256-271.
[68] MATYJASZEWSKI K, PINTAUER T, GAYNOR S. Removal of copper-based catalyst in atom transfer radical polymerization using ion exchange resins [J]. Macromolecules, 2000, 33(4): 1476-1478.
[69] HADDLETON D M, JACKSON S G, BON S A F. Copper(Ⅰ)-mediated living radical polymerization under fluorous biphasic conditions [J]. Journal of the American Chemical Society, 2000, 122(7): 1542-1543.
[70] PAN J, ZHANG B, JIANG X, et al. Cu(Ⅱ)-mediated atom transfer radical polymerization of methyl methacrylate via a strategy of thermo-regulated phase-separable catalysis in a liquid/liquid biphasic system: Homogeneous catalysis, facile heterogeneous separation, and recycling [J]. Macromollecule Rapid Communication, 2014, 35: 1615-1621.
[71] SARBU T, PINTAUER T, MC KENZIE B, et al. Atom transfer radical polymerization of styrenein toluene/water mixtures [J]. Journal of Polymer Science Part A: Polymer Chemitry, 2002, 40: 3153-3160.
[72] HADDLETON D M, DUNCALF D J, KUKULJ D, et al. 3-Aminopropyl silica supported living radical polymerization of methyl methacrylate: Dichlorotris (triphenylphosphine) ruthenium(Ⅱ) mediated atom transfer polymerization [J]. Macromolecules, 1999, 32(15): 4769-4775.
[73] SHEN Y Q, TANG H D, DING S J. Catalyst separation in atom transfer radical polymerization [J]. Progress in Polymer Science, 2004, 29: 1053-1078.
[74] (a) UCHIIKE C, TERASHIMA T, OUCHI M, et al. Evolution of iron catalysts for effective living radical polymerization: Design of phosphine/halogen Ligands in FeX2(PR3)2[J]. Macromolecules, 2007, 40 (24): 8658-8662; (b) UCHIIKE C, OUCHI M, ANDO T, et al. Evolution of Iron catalysts for effective living radical polymerization: P-N chelate ligand for enhancement of catalytic performances [J]. Journal of Polymer Science Part A: Polymer Chemitry, 2008, 46 (20): 6819-6827.
[75] (a) ISHIO M, KATSUBE M, OUCHI M, et al. Active, versatile, and removable iron catalysts with phosphazenium salts for living radical polymerization of methacrylates [J]. Macromolecules, 2009, 42 (1): 188-193; (b) ISHIO M, TERASHIMA T, OUCHI M, et al. Carbonylphosphine hetero-ligated half-metallocene iron(Ⅱ) catalysts for living radical polymerization: Concomitant activity and stability [J]. Polymer Journal, 2010, 42 (1): 17-24; (c) ISHIO M, TERASHIMA T, OUCHI M, et al. Carbonyl phosphine heteroligation for pentamethylcyclopentadienyl (Cp*)Iron complexes: Highly active and versatile catalysts for living radical polymerization [J]. Macromolecules, 2010, 43 (2): 920-926; (d) ISHIO M, OUCHI M, SAWAMOTO M. Dicarbonyl pentaphenylcyclopentadienyl iron complex for living radical polymerization: Smooth generation of real active catalysts collaborating with phosphine ligand [J]. Journal of Polymer Science Part A: Polymer Chemitry, 2011, 49 (2): 537-544.
[76] NISHIZAWA K, OUCHI M, SAWAMOTO M. Phosphine-ligand decoration toward active and robust Iron catalysts in LRP [J]. Macromolecules, 2013, 46 (9): 3342-3349.
[77] (a) TEODORESCU M, GAYNOR S G, MATYJASZEWSKI K. Halide anions as ligands in iron-mediated atom transfer radical polymerization[J]. Macromolecules, 2000, 33 (7): 2335-2339; (b) WANG Y, MATYJASZEWSKI K. ATRP of MMA in polar solvents catalyzed by FeBr2without additional ligand [J]. Macromolecules, 2010, 43(9): 4003-4005.
[78] XUE Z, LINH N T B, NOH S K, et al. Phosphoruscontaining ligands for Iron(III)-catalyzed atom transfer radical polymerization [J]. Angewandte Chemie International Edition, 2008, 47(34): 6426-6429.
[79] WANG Y, KWAK Y, MATYJASZEWSKI K. Enhanced activity of ATRP Fe catalysts with phosphines containing electron donating groups[J]. Macromolecules, 2012, 45(15): 5911-5915.
[80] FUJIMURA K, OUCHI M, SAWAMOTO M. Ferrocene cocatalysis for iron-catalyzed living radical polymerization: Active, robust, and sustainable system under concerted catalysis by two iron complexes [J]. Macromolecules, 2015, 48(13): 4294-4300.
[81] MAGENAU A J D, STRANDWITZ N C, GENNARO A, et al. Electrochemically mediated atom transfer radical polymerization [J]. Science, 2011, 332: 81-84.
[82] OTSU T, KURIYAMA A. Living mono-and biradical polymerizations in homogeneous system synthesis of AB and ABA type block copolymers [J]. Polymer Bulletin, 1984, 11(2): 135-142.
[83] ZHANG W, ZHU X, ZHU J, et al. Atom transfer radical polymerization of styrene using the novel initiator ethyl 2-N, N-(diethylamino)dithiocarbamoylbutyrate [J]. Journal of Polymer Science Part A: Polymer Chemitry, 2005, 44: 32-41.
[84] ZHANG W, ZHOU N, ZHU J, et al. Synthesis of well-defined naphthalene and photo-labile group-labeled polystyrene via ATRP [J]. Journal of Polymer Science Part A: Polymer Chemitry, 2005, 44: 510-518.
[85] DETREMBLEUR C, VERSACE D L, PIETTE Y, et al. Synthetic and mechanistic inputs of photochemistry into the bis-acetylacetonatocobalt-mediated radical polymerization ofn-butyl acrylate and vinyl acetate [J]. Polymer Chemistry, 2012, 3: 1856-1866.
[86] LU L, YANG N, CAI Y. Well-controlled reversible addition-fragmentation chain transfer radical polymerisation under ultraviolet radiation at ambient temperature [J]. Chemical Communications, 2005: 5287-5288.
[87] SHANMUGAM S, XU J, BOYER C. Photoinduced electron transfer-reversible addition-fragmentation chain transfer (PET-RAFT) polymerization of vinyl acetate and N-vinylpyrrolidinone: kinetic and oxygen tolerance study [J]. Macromolecules, 2014, 47(15): 4930-4942.
[88] GUAN Z, SMART B. A remarkable visible light effect on atom-transfer radical polymerization [J]. Macromolecules, 2000, 33(18): 6904-6906.
[89] TASDELEN M A, UYGUN M, YAGCI Y. Photoinduced controlled radical polymerization [J]. Macromolecule Rapid Communication, 2011, 32: 58-62.
[90] TASDELEN M A, UYGUN M, YAGCI Y. Photoinduced controlled radical polymerization in methanol [J]. Macromolecule Chemistry Physics, 2010, 211: 2271-2275.
[91] RIBELLI T G, KONKOLEWICZ D, BERNHARD S, et al. How are radicals(re)generated in photochemical ATRP? [J]. Journal of the American Chemical Society, 2014, 136(32): 13303-13312.
[92] KONKOLEWICZ D, SCHRODER K, BUBACK J, et al. Visible light and sunlight photoinduced ATRP with ppm of Cu catalyst [J]. ACS Macro Letter, 2012, 1(10): 1219-1223.
[93] MOSNACEK J, ILCíKOVA M. Photochemically mediated atom transfer radical polymerization of methyl methacrylate using ppm amounts of catalyst [J]. Macromolecules, 2012, 45(15): 5859-5865.
[94] TREAT N J, FORS B P, KRAMER J W, et al. Controlled radical polymerization of acrylates regulated by visible light [J]. ACS Macro Letter, 2014, 3(6): 580-584.
[95] PAN X, MALHOTRA N, SIMAKOVA A, et al. Photoinduced atom transfer radical polymerization with ppm-level Cu catalyst by visible light in aqueous media [J]. Journal of the American Chemical Society, 2015, 137(49): 15430-15433.
[96] TREAT N J, SPRAFKE H, KRAMER J W, et al. Metal-free atom transfer radical polymerization [J]. Journal of the American Chemical Society, 2014, 136(45): 16096-16101.
[97] PAN X, LAMSON M, YAN J, et al. Photoinduced metal-free atom transfer radical polymerization of acrylonitrile [J]. ACS Macro Letter, 2015, 4(2): 192-196.
[98] MIYAKE G M, THERIOT J C. Perylene as an organic photocatalyst for the radical polymerization of functionalized vinyl monomers through oxidative quenching with alkyl bromides and visible light [J]. Macromolecules, 2014, 47(23): 8255-8261.
[99] LIU X, ZHANG L, CHENG Z, et al. Metal-free photoinduced electron transfer-atom transfer radical polymerization (PET-ATRP) via a visible light organic photocatalyst [J]. Polymer Chemistry, 2016, 7: 689-700.
[100] JORDAN C T, CHERN-HOOI L, YANG H S, et al. Organocatalyzed atom transfer radical polymerization dri-ven by visible light [J]. Science, 2016, 352: 1082-1086.
[责任编辑:吴文鹏]
Progress in atom transfer radical polymerization
CHAI Yun, SONG Yifan, REN Yanrong, ZHOU Hui*
(CollegeofChemistryandChemicalEngineering,InstituteofFineChemistryandChemicalEngineering,EngineeringLaboratoryofFlameRetardantandFunctionalMaterialsofHenanProvince,HenanUniversity,Kaifeng475004,Henan,China)
Atom transfer radical polymerization (ATRP), as a new type of controllable/living polymerization reaction has been developed rapidly. This polymerization technology has been widely used in the molecular structure design of polymers and the synthesis of many functional polymer materials. In this paper, the basis of the reaction mechanism of ATRP is reviewed. The influence of initiator, catalyst, ligand, monomer on ATRP was introduced. The green and efficient reduction of metal salt content were reviewed, such as initiators for continuous activator regeneration ATRP, activators (re)generated by electron transfer for ATRP, ATRP catalysted by iron compond, photo, etc. The highlight of recent development of metal free organic catalyzed ATRP polymerization system were also reviewed.
atom transfer radical polymerization (ATRP); organocatalyzed ATRP; photoinduced; living polymerization
2016-11-17.
国家自然科学基金项目(51203043).
柴 云(1972-),女,实验师,研究方向为功能高分子.*
,E-mail:zhouhui@henu.edu.cn.
O63
A
1008-1011(2017)03-0269-20
猜你喜欢
无机化学学报(2020年7期)2020-07-20
山东化工(2019年13期)2019-02-16
无机化学学报(2018年8期)2018-08-01
中国军转民(2017年7期)2017-12-19
中国塑料(2015年5期)2015-10-14
世界热带农业信息(2014年11期)2015-01-05
中国卫生(2014年10期)2014-11-12
中国神经精神疾病杂志(2014年1期)2014-03-01
中成药(2014年11期)2014-02-28
中国氯碱(2014年11期)2014-02-28